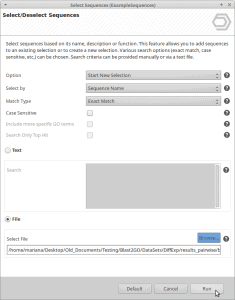
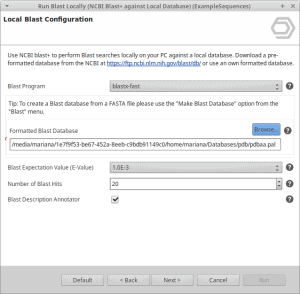
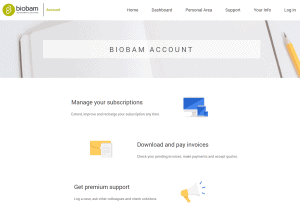
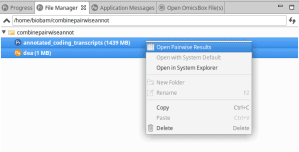
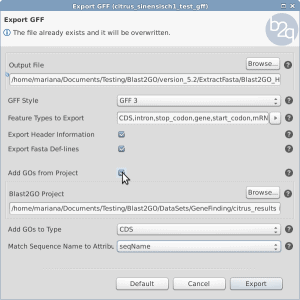
Unlink an OmicsBox subscription from all associated computers
If you reached the maximum number of computers linked to your subscription, this option allows you to unlink all the computers linked to your subscription and to activate new computers. Please note, only the person associated to the subscription can unlink the computers via the BioBam Account. If you logged in to your account and can not see your subscription