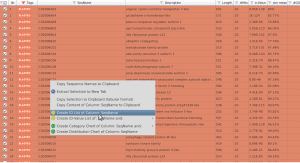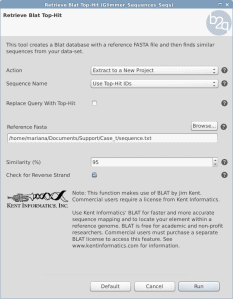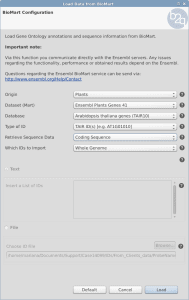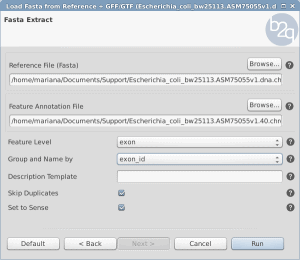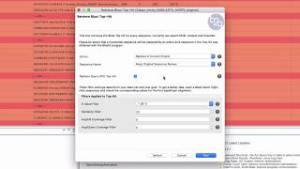
Improve functional annotations using “Retrieve Blast Top-hit”
RNA-seq data is sometimes difficult to match with proteins, due to the short length of the reads. When this is the case, it might be useful to try to find EST hits, which can then be used to find new protein matches. In this demo, we will show how to retrieve top EST hits and the different options that this tool