Create a taxonomic mapping file to Make Blast Database within OmicsBox
OmicsBox allows creating a custom database to run Blast locally. The blast algorithm will run on the user’s computer against a database that is installed locally.
In order to do so, we have to either download a pre-formatted NCBI database (see tutorial) or format our own database (see this tutorial until step 3). When formatting own database an optional file may be used to associate each sequence with a taxonomic node.
In the following use cases we will explain how to create the Taxonomy ID file.
The taxonomic mapping file is a tab delimited text file and should be provided in the following format:
<SequenceId> \t <TaxonomyId><newline>
Use case 1
In this use case we will show how to create a taxonomy mapping file from a fasta file that has been downloaded from NCBI.
Considering we have few sequences e.g. 20 and the taxonomic mapping file can be created by “hand”.
Here are the first 4 sequences from fasta file that we will use as an example.
>gi|513846619|ref|WP_016515408.1| alpha/beta fold hydrolase [Microcystis aeruginosa]
MQQILGLETQPKIWHWRGFKITYQSAGETGPAIVLVHGFGASWGHWRKNIPVLGEKCRCFALDLIGFGGS
DKPEPKNEIDYTFETWGAQIADFCREVVGGAAFLAGNSIGCIAIMQAAVDHPDFVLGVAAINCSLRLLHE
RKRGELPWYRRLGADFAQIILKNKAIGAFFFQQIAKPQTVRKILLQAYRRSEAVTEELVEIILKPARDPG
AIEVFLAFTGYSGGPLPEDLLPILLCPAILLWGSEDPWEPLPLGQELARFPTVKKFIPLAGLGHCPQDEA
PEIVNPILLDFLQAYS
>gi|505170323|ref|WP_015357425.1| alpha/beta fold hydrolase [Mycobacterium liflandii]
MTVTQSKTAEHTFESTSRYAEVDVDGPLKLHYHEAGVGNDQTVVLLHGGGPGASSWSNFARNIEVLAQQF
HVLAVDQPGYGHSDKRAEHGQFNHYAARALKELFDQLGLGRVPLVGNSLGGGTAVRFALDYPDRAGRLVL
MGPGGLSINLFAPDPTEGVKRLGKFSVAPTRENLEAFLRVMVYDQKLITPELVDQRFELACTPESLAATR
AMGKSFAAADFELGMMWREVYKLRQPVLLIWGREDRVNPLDGALVALKTIPRAQLHVFGQCGHWAQVEKF
DEFNRLTIDFLGGAR
>gi|505107425|ref|WP_015294527.1| alpha/beta fold hydrolase [Mycobacterium canettii]
MTATEELTFESTSRFAEVDVDGPLKLHYHEAGVGNDQTVVLLHGGGPGAASWTNFSRNIAVLARHFHVLA
VDQPGYGHSDKRAEHGQFNRYAAMALKGLFDQLGLGRVPLVGNSLGGGTAVRFALDYPDRAGRLVLMGPG
GLSINLFAPDPTEGVKRLSKFSVAPTRENLEAFLRVMVYDKNLITPELVDQRFALASTPESLTATRAMGK
SFAGADFEAGMMWREVYRLRQPVLLIWGREDRVNPLDGALVALKTIPRAQLHVFGQCGHWVQVEKFDEFN
KLTIEFLGGGR
>gi|504907150|ref|WP_015094252.1| alpha/beta fold hydrolase [Pseudomonas sp. UW4]
MGALAQGHFVTLPDGLQLRYVDTGGKDGGNGEPVIFIHGSGPGASGHSNFKQNYTVFAEAGYRVIVPDLP
GYGASDKPDTLYTLDFFVAALSGLLDALDIQRCVLVGNSLGGAIAIKLALDQPQRVSRLVLMAPGGLMEK
EQYYLQMEGIQKMGAAFAKGELNDAAGMRRLLALQLFDESLISDETVNERVAVVQQQPVCVLSTMQVPNM
TSRLGELQCPILGFWGMNDKFCPSSGARTMLENCSRIRFVMLSECGHWVQVEHRDYFNSQCLAFLQEARG
-
The sequence identifier (gi or Acc) (gi|513846619|ref|WP_016515408.1|) can be extracted from the fasta file with the following linux command line. We will extract the Acc identifier in this example.
grep ">" 1_Sequence.txt | cut -d \| -f4 > sequence_name.txt
The sequence_names.txt file looks like:
WP_016515408.1
WP_015357425.1
WP_015294527.1
WP_015094252.1 -
The taxonomic names ([Microcystis aeruginosa]) for each sequence can also be retrieved from the fasta file
grep ">" my_fasta_file.fasta | cut -d '>' -f2 | cut -d '[' -f2 | cut -d ']' -f1 > taxonomy_names.txt
The taxonomy_names.txt file looks like:
Microcystis aeruginosa
Mycobacterium liflandii
Mycobacterium canettii
Pseudomonas sp. UW4 -
Now that you have the taxonomic names you may search for its id in NCBI and create the taxonomy mapping file.
Here is a tool that helps you retrieving the corresponding taxonomic identifiers by giving the taxonomy names: https://www.ncbi.nlm.nih.gov/Taxonomy/TaxIdentifier/tax_identifier.cgi -
The final tab delimited taxonomy ID file, that can be easily created with Excel should look like:
WP_016515408.1 1126
WP_015357425.1 261524
WP_015294527.1 78331
WP_015094252.1 1207075
Copying and pasting data from different files is error prone. The next use case explains how to create a taxonomy ID file directly from a pre-formatted database from NCBI.
Use case 2
Here the main goal is to obtain a class specific database from a pre-formatted database from NCBI.
In this example we want to create a EST databse for class Hydrozoa using an Acc list retrieved from NCBI.
Once we have these files we will create the taxonomic mapping file that can then be used with Make Blast Database feature within OmicsBox.
- Download the Blast+ executables (ftp://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/LATEST/) from NCBI and extract it.
- Download est_others database (with perl script or directly from NCBI webpage)
- At the moment (July 2019) there are 11 files est_others.00.tar.gz to est_others.10.tar.gz
- At the moment (July 2019) there are 11 files est_others.00.tar.gz to est_others.10.tar.gz
- Search the Entrez Nucleotide database and query for a taxonomic identifier e.g. “txid6074[Organism] “
- Download the ACC list from the desired species from NCBI
Send to > Complete Record > File > Accession List
- The above-mentioned Perl script is located in the bin folder of the Blast+ executables and it can be used to download the desired pre-formatted database, e.g. est_others.
To run the script, Perl has to be installed on the computer.perl update_blastdb.pl est_others - Once all .tar.gz files have been downloaded, they need to to be extracted. It is possible to do so in one go with a Linux command.
for file in *.tar.gz; do tar -xvzf $file; done -
Now that we have the .nal database and the Acc list it is possible to create the so called taxonomic mapping file to be used in the create database feature in OmicsBox.
blastdbcmd -db est_others -entry_batch sequence_acclist.txt -outfmt '%i %T' > sequenceID_taxa.txt
- Now you will end up with a text file that has the sequence id and the corresponding taxonomy. This file can be opened with Excel and it contains the | as separator.
emb|HE983324.1| 6087
emb|HE983323.1| 6087
emb|HE983322.1| 6087
emb|AM755036.1| 252671
emb|AM755035.1| 252671
emb|AM755034.1| 252671If it has been opened correctly in Excel then we will end up with 3 columns where the 2nd and 3rd are the ones we need to proceed with the generation of the EST database of the desired species
HE983324.1 6087
HE983323.1 6087
HE983322.1 6087
AM755036.1 252671
AM755035.1 252671
AM755034.1 252671 - To create the desired database within OmicsBox (Functional Analysis > Blast > Make Blast Database) we also need the EST sequences itself in fasta format.
./blastdbcmd -db est_others -entry_batch sequence_acclist.txt -out est_species_6074.fasta
- That is all. You can now select the EST fasta file and the corresponding TaxonomyID file in the Make Blast Database dialog in OmicsBox.
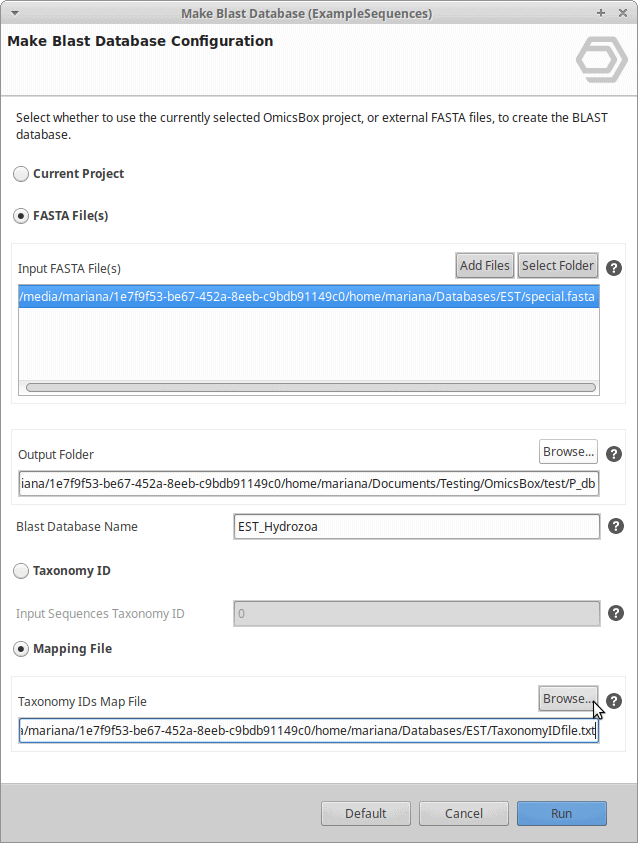
Select .fasta and .txt files to Make Blast Database
For more information on how to create own database or run Local Blast within OmicsBox, please have a look at our user manual, tutorials and videos.

