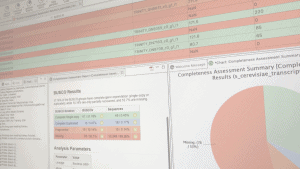
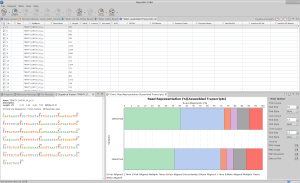
Predict coding regions within transcripts in OmicsBox with TransDecoder
Most transcripts assembled from eukaryotic and prokaryotic RNA-Seq data are expected to code for proteins. The most practical procedure to identify likely coding transcripts is a sequence homology search, such as by BLASTX, against sequences from well-annotated and related species. Predicting coding regions is crucial to determine the molecular role that transcripts play in the cell. Unfortunately, such well-annotated nearby