DNA sequencing is the process of determining the nucleic acid sequence in DNA, and it is the technology by which the genome of a species can be characterized. Despite the advent of next-generation sequencing, current DNA sequencing technologies cannot read whole genomes at once, but rather reads small pieces of between 20 and 30.000 bases, depending on the technology used. To reconstruct the original sequence, sequence assembly techniques are needed.
Genome assembly refers to the bioinformatic process of taking a large number of DNA fragments that are generated during sequencing and assembling them into the correct order such as to reconstruct the original genome. If the genome of a similar organism has been previously sequenced, the genome of interest can be assembled by comparing it to this known genome. However, if the genome to be assembled is not similar to any other organisms whose genomes have been previously sequenced, de novo genome assembly strategies are preferred.
To date, numerous genome assemblers have been developed. According to the approach they use, assemblers are classified as greedy, overlap-layout consensus (OLC) and Hamiltonian path, and de Bruijn graph and Eulerian path. Furthermore, different assemblers are designed for different sequencing technologies and are tailored for particular needs, such as the assembly of small (bacterial) or large (eukaryotic) genomes.
Here we focus on hybrid assembly strategies, such as the one offered in the Genome Analysis module of OmicsBox. In bioinformatics, hybrid genome assembly refers to utilizing various sequencing technologies to achieve the task of assembling a genome from fragmented, sequenced DNA resulting from shotgun sequencing. Usually, hybrid approaches involve supplementing short, accurate second-generation sequencing data (i.e. Illumina or IonTorrent) with long less accurate third-generation sequencing data (i.e. PacBio or Oxford Nanopore) to resolve complex repeated DNA segments. Supplementation of third-generation reads with short, high-accuracy second-generation sequences can overcome inherent errors and completed crucial details of the genome.
The hybrid genome assembly option offered by OmicsBox is based on SPAdes. SPAdes is a de novo genome assembly pipeline that can deal with data coming from several sequencing technologies and supports hybrid and single-cell assemblies. The SPAdes assembly pipeline consists of four stages:
- Assembly graph construction: SPAdes uses the multisized de Bruijn graph, implements new bulge/tip removal algorithms, detects and removes chimeric reads, aggregates biread information into distance histograms, and allows to backtrack the performed graph operations.
- k-bimer adjustment: SPAdes derives accurate distance estimates between k-mers in the genome using joint analysis of distance histograms and paths in the assembly graph.
- Constructs the paired assembly graph: Inspired by Paired de Bruijn graphs (PDBG) approach.
- Contig construction: SPAde constructs DNA sequences of contigs and the mapping of reads to contigs by backtracking graph simplifications.
Hybrid approaches involve supplementing short, accurate second-generation sequencing data with long less accurate third-generation sequencing data to resolve complex repeated DNA segments.
The SPAdes genome assembler is integrated in the Genome Analysis module and can be accessed through the DNA-Seq de novo Assembly option. The first step is to provide the input sequencing data to be provided. SPAdes requires at least one type of short sequencing library, Illumina or Ion Torrent, and it supports single-end, paired-end, and mate-pair data. SPAdes developers advise against assembling both type of libraries (Illumina and IonTorrent) together. In the case of Multiple Displacement Amplification (MDA) single-cell data assembly, the corresponding option should be specified. In addition to these mandatory input data, sequences from other technologies can be provided, such as the produced by PacBio, Oxford Nanopore and Sanger technologies, and contigs of the same genome generated by another assembler.
In addition to the input data, different SPAdes parameters can be configured. The k-mer size can be selected automatically based on read length and data set type, or it can be established by users. SPAdes offers a read error correction step before assembly, recommended to obtain high-quality assemblies. Furthermore, users can specify that SPAdes tries to reduce the number of mismatches and short indels, and establish a minim read coverage to obtain the most reliable assembled sequences.
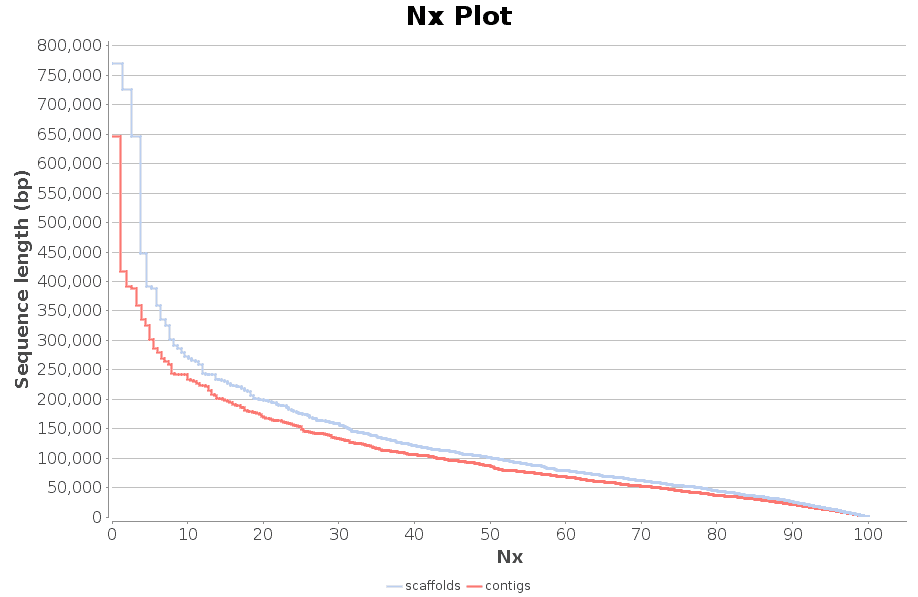
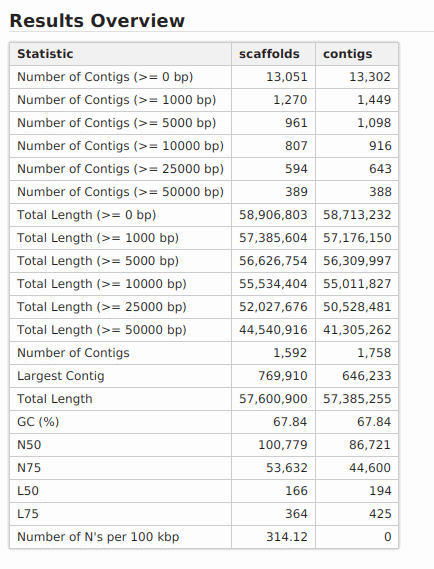
Once finished, the assembled sequences are returned in two different FASTA files, containing contigs and scaffolds. Contigs/scaffolds names in SPAdes output FASTA files contain useful information, such as the sequence length and the k-mer coverage. In addition to the resulting FASTA files, a report and a chart are generated containing a number of common statistics used to describe the quality of a sequence assembly.
References
- Hybrid Genome Assembler User Manual.
- Bankevich A et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. Journal of computational biology : a journal of computational molecular cell biology, 19(5), 455-77.

