High-throughput sequencing of RNA has revolutionized the study of species for which a reference genome is not available or incomplete by enabling the large-scale analysis of their transcriptomes. While analyses of model organisms generally rely on a reference genome, studies of non-model organisms usually lack this advantage. In the absence of an appropriate reference genome, de novo transcriptome assembly is performed.
In this article, we discuss the importance of de novo transcriptome assembly, how to do it and what tools exist for it.
“High-throughput sequencing of RNA has revolutionized the study of species for which reference genome is not available or is incomplete”
What is an RNA-seq de novo assembly and how to perform it with OmicsBox
The aim of de novo transcriptome assembly is to accurately reconstruct the complete set of transcripts that are represented in the read data without the aid of genome sequence information. The resulting assemblies provide the primary data to identify all expressed transcripts, to discover isoforms and facilitate the quantitative assessment of differential gene expression.
To perform de novo transcriptome assembly it is necessary to have a specific tool for it. One of the main functionalities of Blast2GO is RNA-Seq de novo assembly and it is based on the well-known Trinity assembler software developed at the Broad Institute and the Hebrew University of Jerusalem.
Trinity represents a novel method for the efficient and robust de novo reconstruction of transcriptomes from RNA-Seq data. It divides the RNA sequence data into several individual de Bruijn graphs, each representing the transcriptional complexity at a given gene or locus, and then processes each graph independently to extract full-length splicing isoforms and to tease apart transcripts derived from paralogous genes.
“One of the main functionalities of OmicsBox is RNA-Seq de novo assembly and it is based on the well-known Trinity assembler software”
To perform an RNA-seq de novo assembly in OmicsBox it is necessary to open the wizard and to provide the following information:
- Sequence Data: Provide the FASTQ files containing the RNA-sequencing reads. Both, single-end and paired-end data are supported.
- Strand Specificity: Point out if the RNA-seq data comes from strand-specific sequencing protocols.
- Minimizing Falsely fused Transcripts: In case of gene-dense compact genomes, like those of some fungi species, fusion transcripts can be minimized.
- Pair Distance: For Paired-end data, specifies the maximum length expected between fragment pairs.
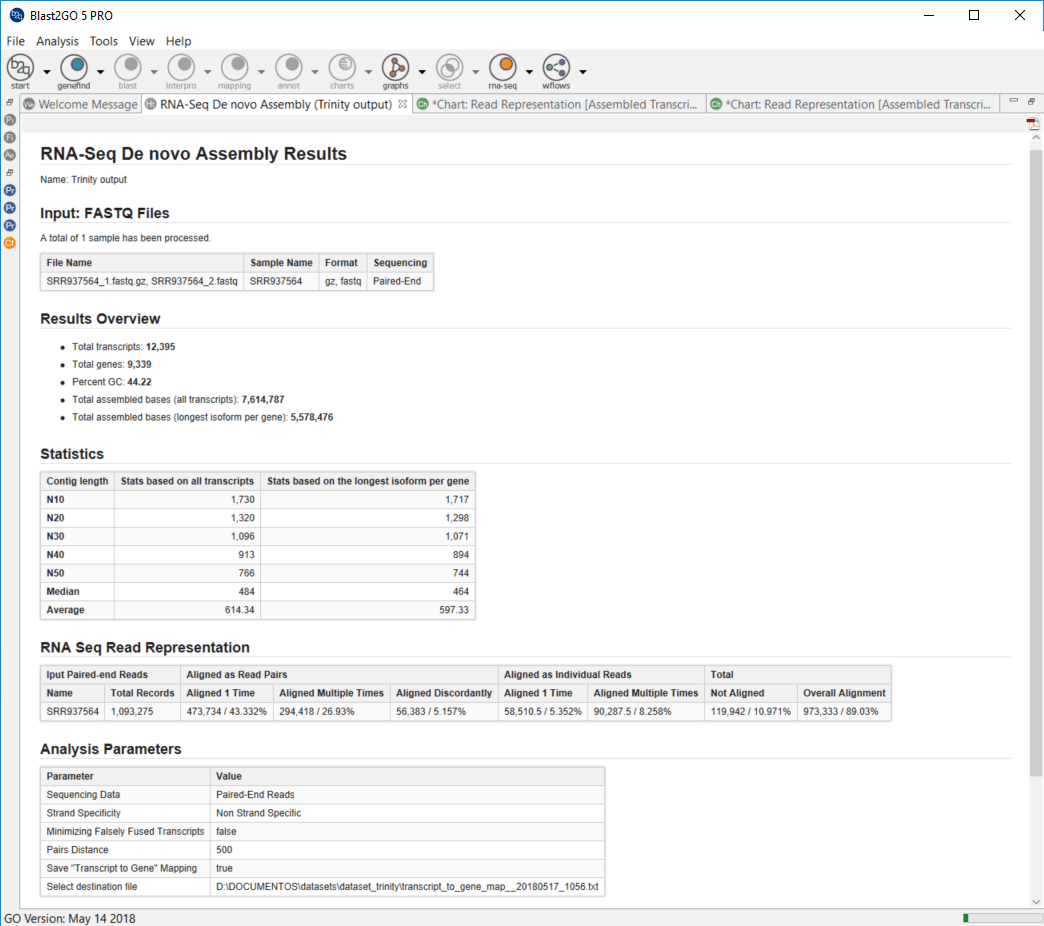
When the RNA-seq de novo assembly completes, a project containing the assembled transcript sequences is returned. Trinity groups transcripts into clusters based on shared sequence content and this transcript clusters can be considered as genes. Since the Trinity FASTA accession encodes the gene and isoform information, OmicsBox provides a text file containing the relationship between gene and isoforms that can be used for downstream analysis.
Furthermore, a result page shows a summary or the assembly results, such as the number of total transcripts and genes detected, the percentage of GC, the total assembled bases, the Nx length statistics and the read composition of the assembly. This information helps to evaluate the quality of the assembly, as well as to determine if it is complete enough to be used in downstream analysis.
“Trinity groups transcripts into clusters based on shared sequence content and this transcript clusters can be considered as genes”
Evaluation and functional characterization of RNA-seq de novo assembly
Once the assembly is complete, there are several analysis to explore aspects of the biology of the organisms based on the assembled transcripts and the input RNA-seq data. The reconstructed transcripts can be evaluated and characterized taking advantage of different functionalities of OmicsBox Modules.
The next step in a standard transcriptomics study is often the characterization of the molecular functions or pathways in which the reconstructed transcripts are involved. Blast2GO allows massive annotation of complete transcriptome datasets against a variety of databases and controlled vocabulary.
Basically, OmicsBox Functional Analysis Module performs BLAST searches to find similar sequences to input transcript sequences. Then, it extracts the Gene Ontology terms associated with each of the obtained hits and returns an evaluated GO annotation for the query sequences. Enzyme codes are obtained by mapping from equivalent GOs while InterPro motifs are directly queried at the InterProScan web service.
In addition, OmicsBox provides several resources that allow to analyze different aspects of the transcriptome, as well as to obtain biological insights: load pathway-maps from KEEG, Rfam blast searches, coding potential assessment, obtain COG orthologous groups, and more.
As you have seen, OmicsBox is a complete tool to perform de novo transcriptome assembly and evaluated and characterized the results of the study. You can try this functionality and many others by requesting a free Trial of OmicsBox, and if you need further information you can check the user manual.

