Most transcripts assembled from eukaryotic and prokaryotic RNA-Seq data are expected to code for proteins. The most practical procedure to identify likely coding transcripts is a sequence homology search, such as by BLASTX, against sequences from well-annotated and related species. Predicting coding regions is crucial to determine the molecular role that transcripts play in the cell. Unfortunately, such well-annotated nearby species are often not available for transcriptomes of newly sequenced transcriptomes.
When we work with non-model organisms, the reference genome and transcriptome might not be available, so transcriptome assembly requires de novo strategies. These newly targeted transcriptomes generally encode proteins that are insufficiently represented by detectable homologies to known proteins. To capture those coding regions, we need methods that predict coding regions based on metrics tied to sequence composition, such as TransDecoder. This tool to Predict Coding Regions is available in the Transcriptomics Module in OmicsBox.
Predict Coding Regions with TransDecoder
TransDecoder is a utility developed and included with Trinity (which can also be used inside OmicsBox). It is intended to assist in the identification of potential coding regions within reconstructed transcripts. TransDecoder recognizes likely coding sequences based on the following criteria:
-
A minimum length open reading frame (ORF) in a transcript sequence.
-
A log-likelihood score is greater than 0. This score is similar to the GeneID score.
-
The above coding score is greatest when the ORF is in the first reading frame as compared to scores in the other 2 forward reading frames.
-
If a candidate ORF is fully encapsulated by the coordinates of another candidate ORF, it reports the longer one. However, a single transcript can report multiple ORFs (allowing for operons, chimeras, etc).
-
A Position-Specific Scoring Matrix (PSSM) is computed, trained, and used to refine the start codon prediction.
Optionally, to further maximize sensitivity for capturing ORFs that may have functional significance, regardless of the coding likelihood score, ORFs can be scanned for homology to known proteins and retain all such ORFs using PFAM to identify common protein domains.
TransDecoder in OmicsBox
The TransDecoder methodology is available in OmicsBox via the Predict Coding Regions utility. Once Trinity has assembled short reads, the Predict Coding Regions procedure can be easily applied. First, users can adjust the tool according to the data and the species under study. They can select the appropriate genetic code to find ORFs, establish a minimum protein length cutoff, configure how ORFs will be retained, etcetera. The Pfam Search is an optional but highly recommended parameter. If checked, ORFs will be scanned against PFAM to identify protein domains, which will be used as ORF retention criteria.
There are three different formats in which the identified ORFs return:
-
CDSs: Nucleotide sequences for coding regions of the final candidate ORFs, in FASTA format.
-
Proteins: Peptide sequences for the final candidate ORFs, in FASTA format.
-
Coordinates: Positions within the target transcripts of the final selected ORF, in GFF format.
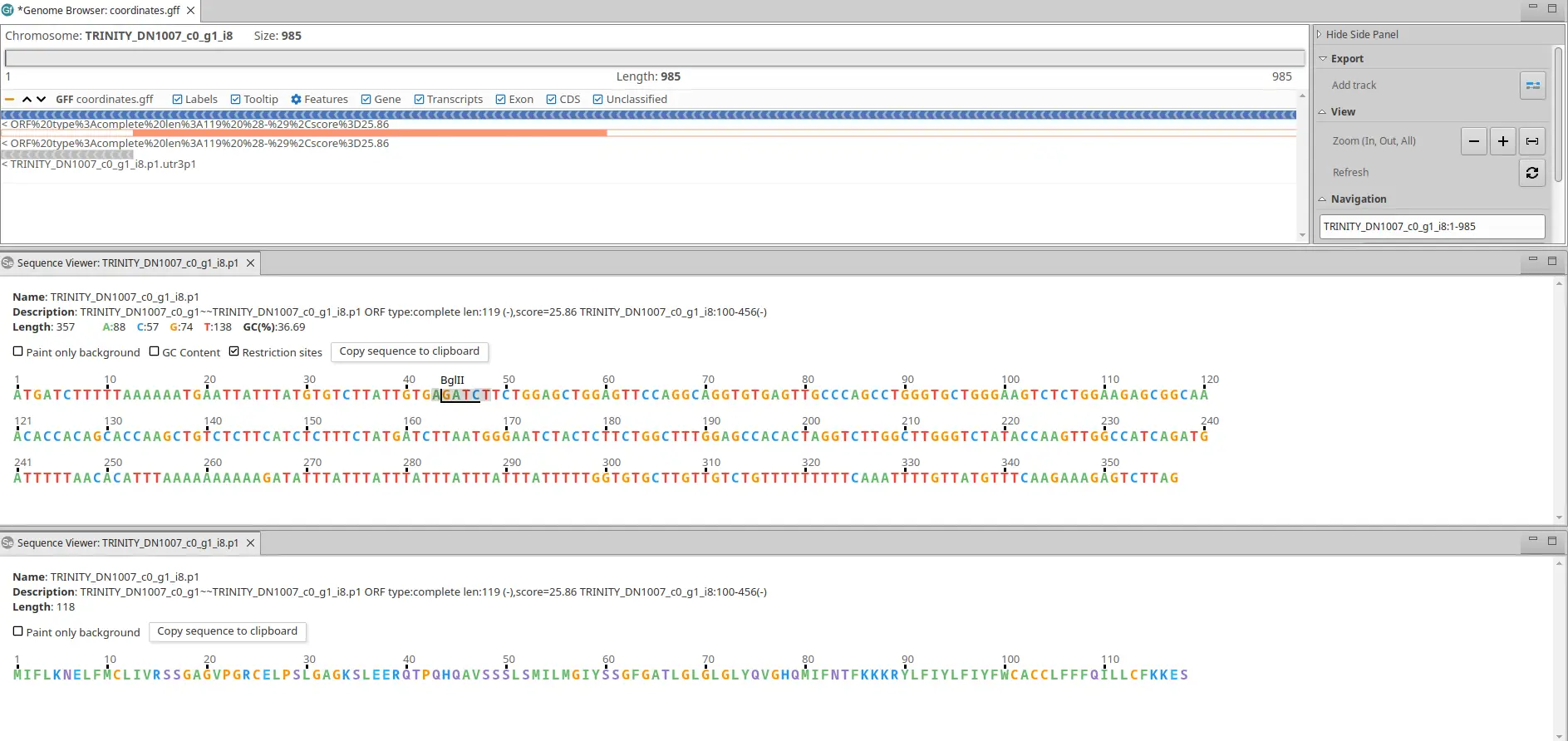
We recommend the OmicsBox Genome Browser for viewing the candidate ORFs in the context of the transcriptome (Fig. 1).
ORF Types
There are a few items to pay attention to in the above files. TransDecoder provides details about the predicted ORFs, such as the length and the strand in which the coding region was found. Furthermore, it classifies predicted ORF according to the start and stop signals (Fig. 2):
-
Complete ORF: Contains a start and a stop codon.
-
5′ partial ORF: It lacks the start codon and presumably part o the N-terminus.
-
3′ partial ORF: It lacks the stop codon and presumably part of the C-terminus.
-
Internal ORF: It is both 5′ and 3′ partial.
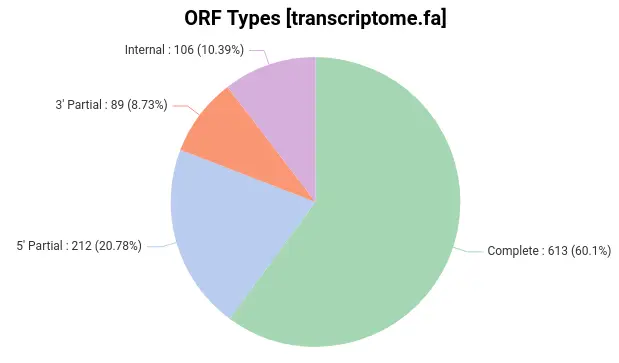
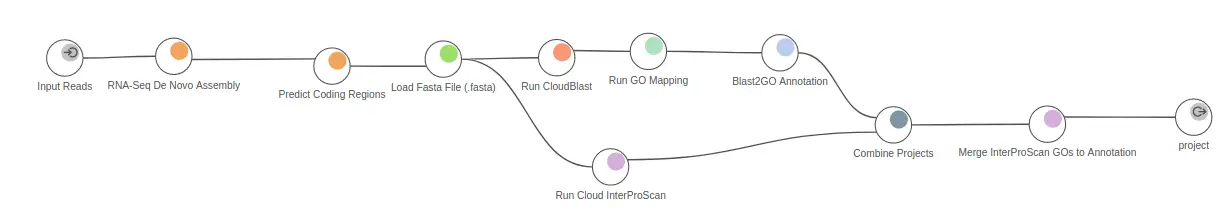
In practice, after applying this strategy, Omicsbox extracts the most confident ORFs, used to predict protein functions. In OmicsBox, this step can be directly linked to the homology-based functional annotation pipeline, which uses the widely known Blas2GO methodology (Fig. 3).
Example Use Case
Reanalyzing the A. galli transcriptomic response to an anthelmintic drug with OmicsBox
References
- Predict Coding Regions User Manual.
- Haas BJ et al. (2013). De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nature protocols, 8(8), 1494-512.
- TransDecoder 5.5.0. Haas, BJ. and Papanicolaou, A. 2019. https://github.com/TransDecoder/TransDecoder/wiki.
About the Author

With a biological and technological academic background, including a BSc in Biotechnology and an MSc in Bioinformatics, Enrique’s expertise lies in the areas of Long Reads and Genetic Variation.

