Defining transcript models to characterize isoforms from aligned reads is a crucial step in analyzing transcriptomics data, particularly when dealing with long reads originating from technologies such as Pacific Biosciences (PacBio) or Oxford Nanopore Technologies (ONT).
In the past few years, so many algorithms to achieve this task have been published, that it is challenging to keep an overview of all of them. Choosing the wrong tool can have grave consequences on downstream analysis results, for instance regarding differential expression analyses. The LRGASP Consortium benchmarking effort made great progress in providing in-depth analysis results to more accurately gauge the capabilities, strengths, and shortcomings of such tools. While its results are by no means conclusive, especially considering the fast-paced developments in the field, their insightful recommendations serve as useful guidelines on which tool to choose for a particular purpose.
While one of their recommendations, FLAIR, has already been available for convenient use in OmicsBox for some time now, we have now implemented a second, alternative tool for this purpose: IsoQuant[1]. To highlight the differences between these two approaches to identifying isoforms, here is a quick overview of their capabilities:
| FLAIR | IsoQuant | |
|---|---|---|
| Strategy | Cluster-based | Graph-based |
| Primary Inputs |
|
|
| Additional Inputs | Reference Annotation AND/OR supporting short reads (at least one of these is required) | Reference Annotation (optional) |
| Outputs |
|
|
| Resources | Repository | Repository |
Based on this (albeit simplified) comparison, we can see that the main difference between these two algorithms lies in their strategy. FLAIR tries to cluster similar reads together and then assembles transcript models based on these clusters. IsoQuant instead uses the reads to construct an intron-graph (see Figure 1). In this graph, the connections between introns are defined by the reads, more specifically their splice junctions. The different paths that wind through these graphs can then be used to define transcript models.
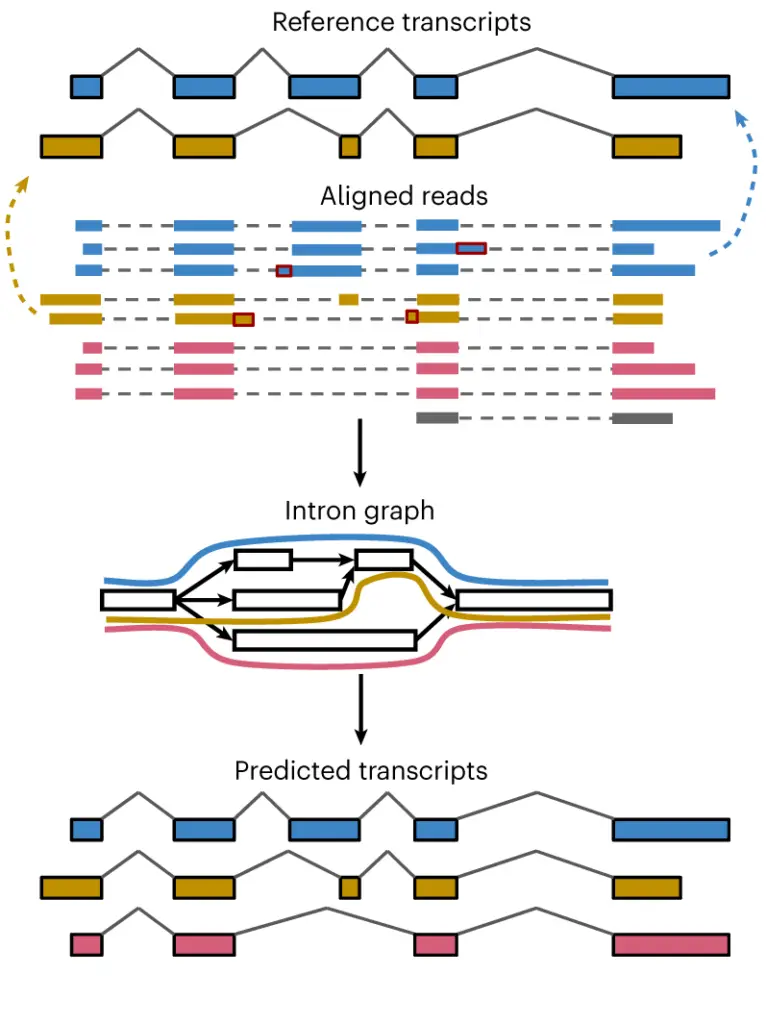
Figure 1: Outline of the IsoQuant pipeline. When providing reference gene annotations, IsoQuant assigns reads to annotated isoforms, and corrects alignment artifacts (top). Then, it constructs an intron graph from read alignments (middle) and discovers transcripts via path construction (bottom).
The time required to run these analyses depends on the size of the data set, though both algorithms are faster than others in the field.
To decide which of these two tools to use, several factors should be considered:
- Though results will always be more reliable when a reference annotation is provided, depending on the data at hand, this is not always possible. When there is no (high-quality) reference annotation available, IsoQuant in “discovery mode” can be a useful option. This mode runs with neither reference annotations, nor supporting short reads.
- When supporting short reads are available, FLAIR may be preferable as it can make use of this orthogonal data to improve the quality of discovered novel transcripts. Specifically, short reads are used to correct splice sites.
- IsoQuant also offers quantification based solely on the reference annotation, without the construction of transcript models. This feature may be of interest if you already have high-quality reference annotations, or want to use a different algorithm to define transcript models. For instance, you could use FLAIR to define a transcriptome, then refine it using SQANTI3 Filter and Rescue, and then use the resulting .gtf file to quantify genes and transcripts with IsoQuant. If you want to perform this reference-based quantification in OmicsBox, remember to uncheck the transcript-model-based outputs on the final page of the IsoQuant wizard to improve computational efficiency.
How to use IsoQuant in OmicsBox
- Introduce Long-Read Data: Import the Long-Read data (as FASTA/Q or BAM format) and the reference genome (as FASTA). If the reads are in FASTA/Q format, IsoQuant will internally run minimap2 to align them to the genome. When working with multiple samples, each sample should be one individual FASTA/Q or BAM file. This way, the quantification step produces a count table with one column per sample.
- (Optional) Introduce Reference Annotations: Optionally, reference annotations in GFF/GTF format can be provided. While this is strongly recommended, when good-quality reference annotations are not available for a species, IsoQuant can also run without reference annotations.
- Configure Algorithm Parameters: Adjust the algorithm parameters to your data set and analysis goals. The different options are explained in detail in the OmicsBox User Manual .
- Configure Outputs: Adjust the output options to ensure you receive the results you are interested in.
- Visualize and Analyze: Explore the outcome of your IsoQuant analysis with the provided report, plot, and tables.
- Proceed with Downstream Analysis: Based on the outputs of IsoQuant, two main avenues of further downstream analysis open up:
- Investigating the identified transcript models: After running IsoQuant, you can use SQANTI3 in OmicsBox to further analyze the resulting transcript models. Among other useful insights, most importantly this allows you to identify the structural categories of your isoforms.
- Performing differential expression analysis: OmicsBox also offers edgeR, NOISeq, and maSigPro as options for differential expression analysis. Which of these to use depends on your experimental setup and the goal of your analysis. Afterward, differentially expressed genes can be connected to the associated Gene Ontology terms through a Gene Set Enrichment Analysis (GSEA) or Fisher’s Exact Test. This step requires a set of Gene Ontology annotations, which can be obtained from a platform such as BioMart, or by running a BLAST.
Conclusion
By running IsoQuant in OmicsBox, you can easily characterize isoforms into transcript models from your Long-Read data. You will benefit from the user-friendly interface and seamless integration of the powerful OmicsCloud infrastructure. Additionally, you can also immediately continue with a downstream analysis of your results, without having to leave OmicsBox!

References
- Prjibelski, A. D., Mikheenko, A., Joglekar, A., Smetanin, A., Jarroux, J., Lapidus, A. L., & Tilgner, H. U. (2023). Accurate isoform discovery with IsoQuant using long reads. Nature Biotechnology, 1-4. https://doi.org/10.1038/s41587-022-01565-y
- Tang, A. D., Soulette, C. M., van Baren, M. J., Hart, K., Hrabeta-Robinson, E., Wu, C. J., & Brooks, A. N. (2020). Full-length transcript characterization of SF3B1 mutation in chronic lymphocytic leukemia reveals downregulation of retained introns. Nature communications, 11(1), 1438. https://doi.org/10.1038/s41467-020-15171-6
- Pardo-Palacios, F. J., Wang, D., Reese, F., Diekhans, M., Carbonell-Sala, S., Williams, B., … & Brooks, A. N. (2021). Systematic assessment of long-read RNA-seq methods for transcript identification and quantification. bioRxiv. https://www.doi.org/10.1101/2023.07.25.550582
- Identificacion and Quantification with IsoQuant, OmicsBox User Manual .
About the Author


