Introduction
Driven by advances in long-read sequencing technologies, high-quality phased (haplotype-resolved) reference genomes are becoming increasingly available for complex polyploid organisms, including agronomically important crops. In this blog post, we will explain what phased reference genomes are and how they can be combined with long-read RNA-seq to unlock the secrets of allele- and haplotype-specific gene expression in complex polyploid organisms.
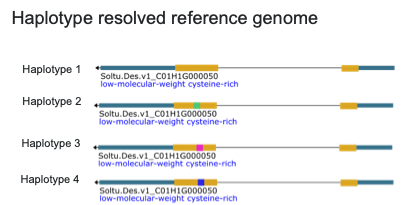
What Are Phased Reference Genomes?
Unlike traditional genome assemblies that collapse sequence variation into a single
consensus, phased reference genomes represent each haplotype as a distinct
sequence. This distinction is crucial for organisms with complex, polyploid genomes,
where multiple copies of each chromosome coexist and can differ substantially at the
sequence level.
Thanks to rapid advances in long-read sequencing technologies, high-quality phased
assemblies are becoming increasingly available for agronomically important crops.
Examples include tetraploid blueberry (1), tetraploid potato (2), hexaploid wheat (3), and
hexaploid sweetpotato (4).
These resources are opening new possibilities for studying gene regulation at the
haplotype level.
Allele-Specific Expression Analysis
What Is Allele-Specific Expression?
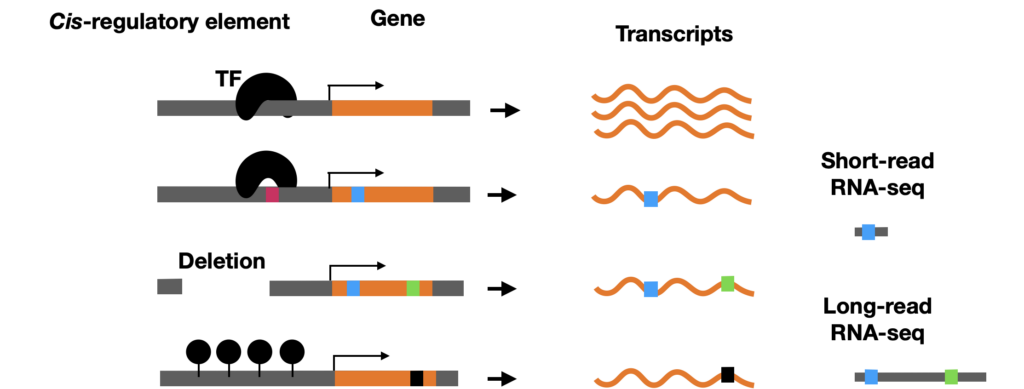
Allele-specific expression (ASE) analysis identifies cis-regulatory differences between haplotypes (5, 6), that is, cases where two copies of the same gene are expressed at different levels due to sequence differences in their regulatory regions rather than trans-acting environmental factors. As illustrated in Figure 2, cis-regulatory variation can arise from differences in promoter sequence, such as SNPs or structural variations.
Why Use Phased Genomes for Allele-specific expression analysis?
Phased reference genomes make Allele-specific expression analysis considerably easier. By providing haplotype-resolved sequences, they allow RNA-seq reads to be assigned back to their haplotype of origin, a step that is unreliable or impossible with a collapsed consensus assembly. This is especially important for resolving complex haplotypes in highly polyploid species, where a single gene can have three or more copies that are difficult to distinguish with short reads alone.
Practical Considerations for dealing with phased reference genomes
Working with phased genomes in an Allele-specific expression framework requires careful attention to several bioinformatic details to avoid bias in the results.
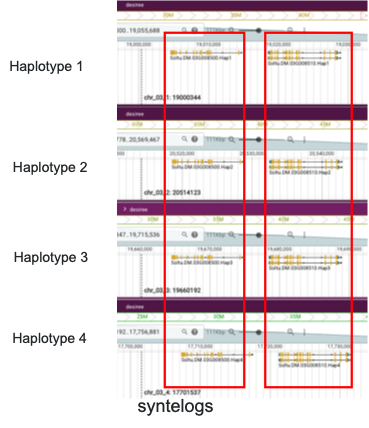
Syntelog identification. Before quantification, conserved genes across haplotypes (syntelogs) must be grouped together. This ensures that expression comparisons are made between true haplotypic counterparts rather than unrelated gene copies. As shown in Figure 3, syntelogs are sets of genes in a phased assembly that are orthologous copies of the same ancestral gene across haplotypes.
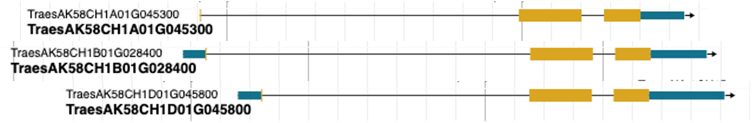
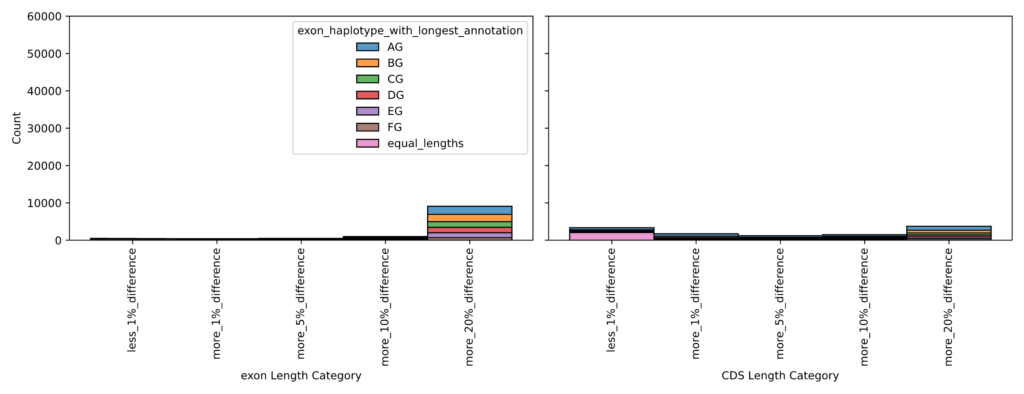
UTR annotation quality. Differences in UTR length between haplotypes can introduce quantification bias (Figure 4). When UTR annotations are unreliable, it is advisable to restrict analysis to CDS coordinates or to use a lifted-over annotation from a well-annotated reference haplotype.
Unphased chromosomes. Phased assemblies frequently contain a mixture of phased and unphased sequences (e.g., Chr06 vs. Chr06A–F). For syntelog identification, it is best to filter to phased chromosomes only; however, all sequences should be retained for read mapping to avoid misassignment of reads from unphased regions.
Organellar genomes. Mitochondrial and chloroplast sequences should always be included in the reference. Reads mapping to organellar genomes must be accounted for to avoid spurious haplotype assignments in the nuclear genome.
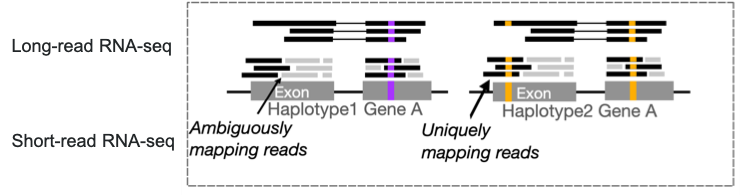
Multimapping and long reads. The high multimapping rate characteristic of short-read RNA-seq in polyploid genomes makes haplotype assignment unreliable. As illustrated in Figure 5, long-read RNA-seq dramatically reduces multimapping rates when mapping to phased reference genomes, as the longer reads span more haplotype-informative variants and can be assigned with much greater confidence. In addition, allows long-read RNA-seq identification of novel isoforms and better isoform quantification.
Use Case: Allele-specific expression in Sweetpotato
To illustrate this workflow, we applied it to hexaploid sweetpotato (2n = 6x = 90) using the Beauregard v4 phased assembly (4). Sweetpotato is a globally important food security crop, and its complex hexaploid genome makes it an excellent test case for haplotype-resolved expression analysis.
Syntelog Identification
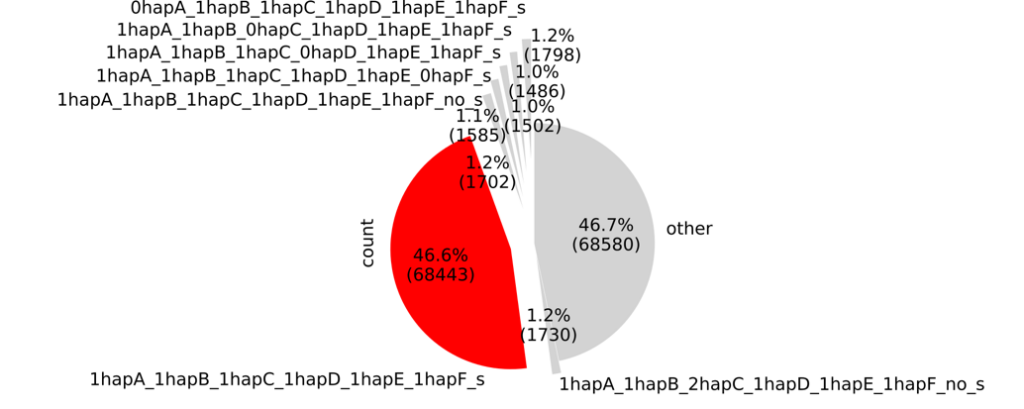
We ran SyntelogFinder (v1.0.0) on the phased assembly via Nextflow and Singularity, to identify the syntelogs that we want to compare for the allele-specific expression analysis. Approximately 50% of coding genes had identifiable syntelogs, a result consistent with observations in potato and other polyploid species (Figure 6).
Reference Preparation and QC
Inspection of the annotation revealed that CDS lengths were consistent across haplotypes, but full transcript lengths varied substantially due to differences in UTR annotations (Figure 7). To avoid quantification bias, CDS features were converted to exon features so that CDS coordinates would be used for expression quantification. The GTF was subsequently validated and repaired with gffread. Organellar genomes (mitochondria and chloroplast) were downloaded from NCBI and appended to both the reference genome and the annotation.
Mapping and Downstream Analysis
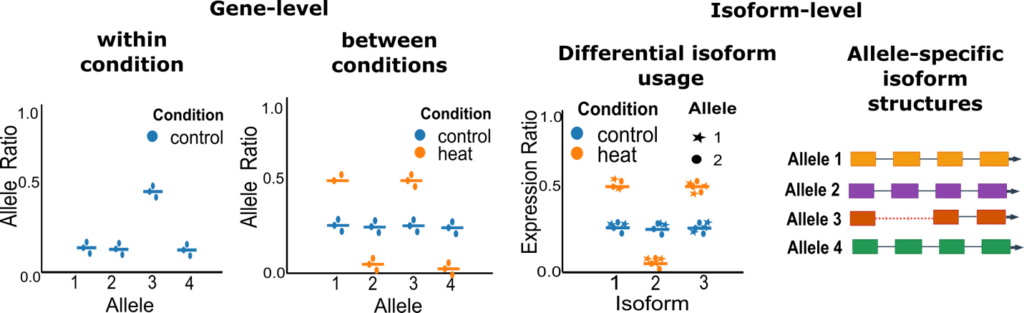
The final reference was fed into the longrnaseq Nextflow pipeline, which includes Bambu (8) for novel isoform discovery, SQANTI3 (9) for quality control and quantification of direct Oxford Nanopore Technology (ONT) long reads with oarfish (10). Allele-specific quantification was then performed using the polyASE Python package (Figure 8), enabling systematic identification differences in allelic expression within conditions and between conditions on the gene level. On the isoform level differential isoform usage and allele-specific isoforms can be identified.
Example Results
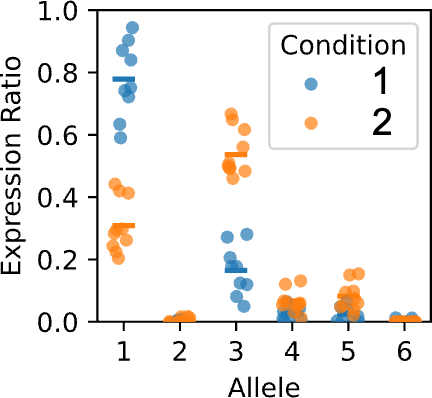
Figure 9 shows an example gene in sweetpotato that exhibits differential allele expression between the two conditions. This is an indication that the alleles respond differently to condition specific transcription factors.
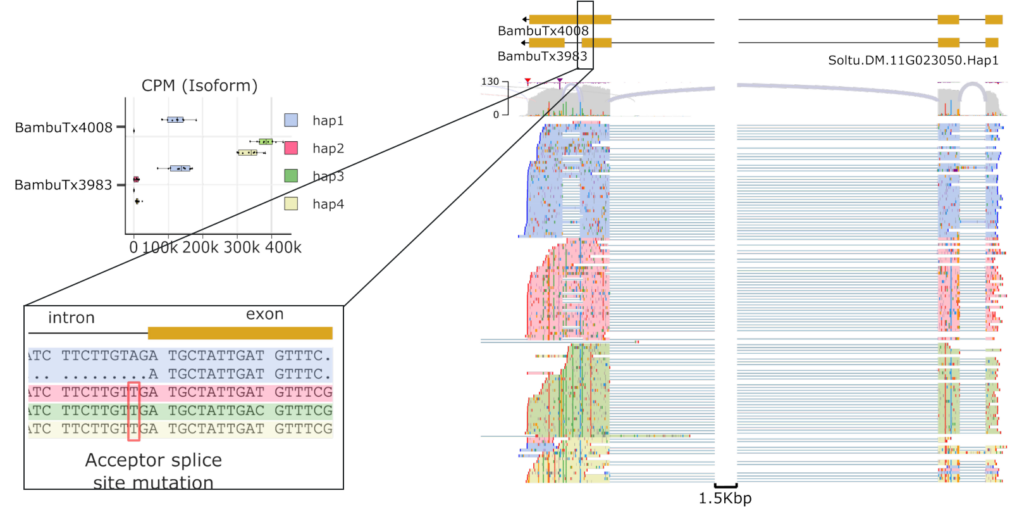
Beyond ASE, the LongPolyASE framework also enables detection of isoform-level differences between haplotypes. Figure 10 shows an example from potato where the dominant isoform differs between haplotypes due to a mutation in a splice site, a finding that would be invisible to short-read or non-haplotype-resolved approaches.
Conclusions
Phased reference genomes, combined with long-read RNA-seq, provide a powerful framework for allele- and isoform-specific expression analysis in polyploid crops. While the workflow requires careful handling of annotation quality, syntelog grouping, and reference construction, the result is a haplotype-resolved view of gene expression that is simply not achievable with short reads and collapsed assemblies. This approach has the potential to identify alleles and isoforms with important roles in crop production, stress response, and agronomic trait variation.
To learn more about the longPolyASE framework, visit the documentation and tutorials and the preprint (5).
References
- Colle,M., Leisner,C.P., Wai,C.M., Ou,S., Bird,K.A., Wang,J., Wisecaver,J.H., Yocca,A.E., Alger,E.I., Tang,H., et al. (2019) Haplotype-phased genome and evolution of phytonutrient pathways of tetraploid blueberry. GigaScience, 8.
- Godec,T., Beier,S., Rodriguez-Granados,N.Y., Sasidharan,R., Abdelhakim,L., Teige,M., Usadel,B., Gruden,K. and Petek,M. (2025) Haplotype-resolved genome assembly of the tetraploid potato cultivar Désirée. Sci Data, 12, 1044.
- Jia,J., Zhao,G., Li,D., Wang,K., Kong,C., Deng,P., Yan,X., Zhang,X., Lu,Z., Xu,S., et al. (2023) Genome resources for the elite bread wheat cultivar Aikang 58 and mining of elite homeologous haplotypes for accelerating wheat improvement. Molecular Plant, 16, 1893–1910.
- Wu,S., Sun,H., Zhao,X., Hamilton,J.P., Mollinari,M., Gesteira,G.D.S., Kitavi,M., Yan,M., Wang,H., Yang,J., et al. (2025) Phased chromosome-level assembly provides insight into the genome architecture of hexaploid sweetpotato. Nat. Plants, 11, 1951–1959.
- Cleary,S. and Seoighe,C. (2021) Perspectives on Allele-Specific Expression. Annu. Rev. Biomed. Data Sci., 4, 101–122.
- Pastinen,T. (2010) Genome-wide allele-specific analysis: insights into regulatory variation. Nat Rev Genet, 11, 533–538.
- Nolte,N., Petek,M., Angulo Lara,P., Mulroney,L., Nicassio,F., Marroni,F. and McIntyre,L. (2025) The promise of long-read RNA-seq: reducing bias in analyses of allele imbalance. 10.1101/2025.10.14.682301.
- Chen,Y., Sim,A., Wan,Y.K., Yeo,K., Lee,J.J.X., Ling,M.H., Love,M.I. and Göke,J. (2023) Context-aware transcript quantification from long-read RNA-seq data with Bambu. Nat Methods, 20, 1187–1195.
- Pardo-Palacios,F.J., Arzalluz-Luque,A., Kondratova,L., Salguero,P., Mestre-Tomás,J., Amorín,R., Estevan-Morió,E., Liu,T., Nanni,A., McIntyre,L., et al. (2024) SQANTI3: curation of long-read transcriptomes for accurate identification of known and novel isoforms. Nat Methods, 21, 793–797.
- Zare Jousheghani,Z., Singh,N.P. and Patro,R. (2025) Oarfish : enhanced probabilistic modeling leads to improved accuracy in long read transcriptome quantification. Bioinformatics, 41, i304–i313.
About the Author & LongTREC

Nadja Nolte
Nadja is a PhD student at the National Institute of Biology in Slovenia, focusing on allele-specific expression analysis in potatoes under heat stress using long-read RNA-seq. As part of the LongTREC Marie Curie PhD network, she had the opportunity to spend two months at BioBam, exchanging ideas about potatoes, explaining that sweetpotatoes are not “real” potatoes, and learning about the differences between industry and academia.

