Introduction
Working with large FASTA and FASTQ files is a routine part of modern bioinformatics, but it is rarely a trivial task. Reference genomes, transcriptome assemblies, protein databases, and high-throughput sequencing reads can easily reach tens of gigabytes, making them difficult to inspect, share, or process on standard hardware. Splitting these files into smaller, more manageable parts is often a necessary first step before running downstream analyses, distributing jobs across a compute cluster, or simply extracting a subset of sequences of interest.
Traditionally, splitting sequence files has required command-line tools and some familiarity with shell scripting or regular expressions. While powerful, this approach can be error-prone and intimidating for users without a strong programming background, especially when dealing with paired-end reads, custom ID patterns, or very large datasets.
This is where the new Split FASTQ/FASTA Files tool in OmicsBox 4.0 comes into play. Powered by SeqKit, a cross-platform, ultrafast toolkit for FASTA/Q manipulation, this new feature brings high-performance sequence splitting into the familiar OmicsBox graphical interface. No command line, no scripting: just a clean wizard that lets you split, group, or extract sequences in a few clicks, whether you are working with a single file or a large batch of mixed FASTA and FASTQ inputs.
How to use the FASTA/FASTQ Splitter in OmicsBox
Setting up the tool
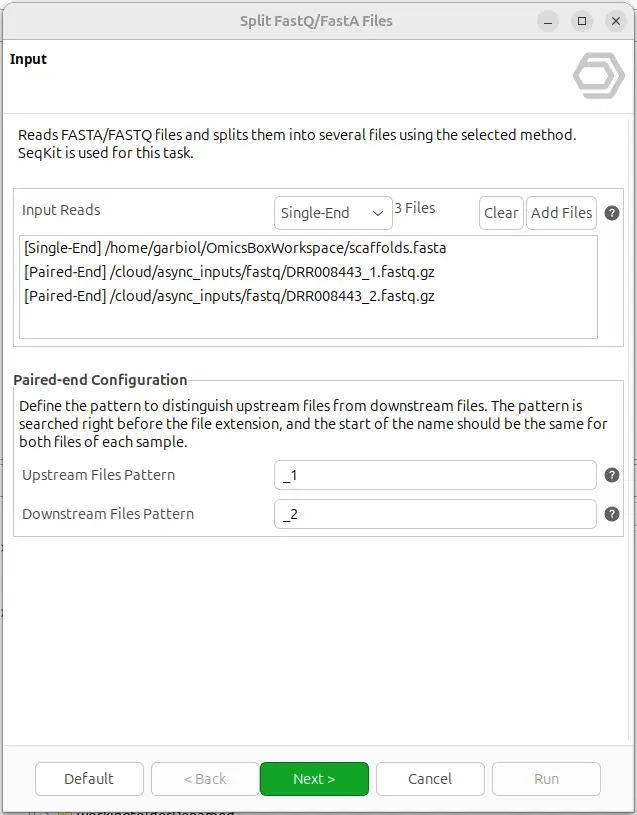
The splitter is available in the General tools module of OmicsBox, under FastQ Tools → Split FastQ/FastA files. The wizard accepts one or more files at once and can process a mix of FASTA and FASTQ inputs in the same run, applying the same splitting operation to all of them.
For each file, the user indicates whether it is single-end or paired-end. The paired-end option is only available for FASTQ files and relies on standard suffix conventions to automatically detect read pairs, so that R1 and R2 are kept consistent across the resulting output files (Figure 1).
Splitting options
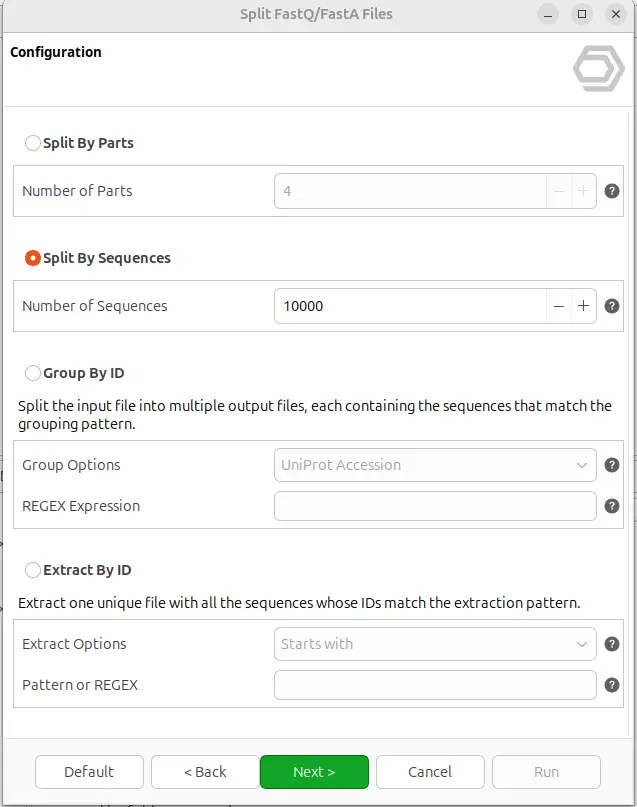
The tool offers four different splitting modes, selectable from the configuration page of the wizard (Figure 2). The appropriate mode depends on the biological question and the downstream analysis:
- Split By Parts: Divides each input file into N equal parts. Useful when you need to parallelize a downstream analysis across several cores or cluster nodes, or simply to break a very large file into chunks of predictable size.
- Split By Sequences: Splits each input file into multiple files with at most N sequences per output. Ideal when a downstream tool has limits on the number of sequences per run, or when you want uniformly sized batches regardless of the total file size.
- Group By ID: Splits each input file into multiple output files, each containing all the sequences whose ID matches a common pattern. This is particularly useful for multi-species FASTA files, UniProt databases, or any dataset where sequences can be naturally grouped by a shared identifier.
- Extract By ID: Produces a single output file containing only the sequences whose ID matches a user-defined pattern. This is the mode to choose when you want to pull a specific subset of sequences out of a large database.
Splitting sequences by ID
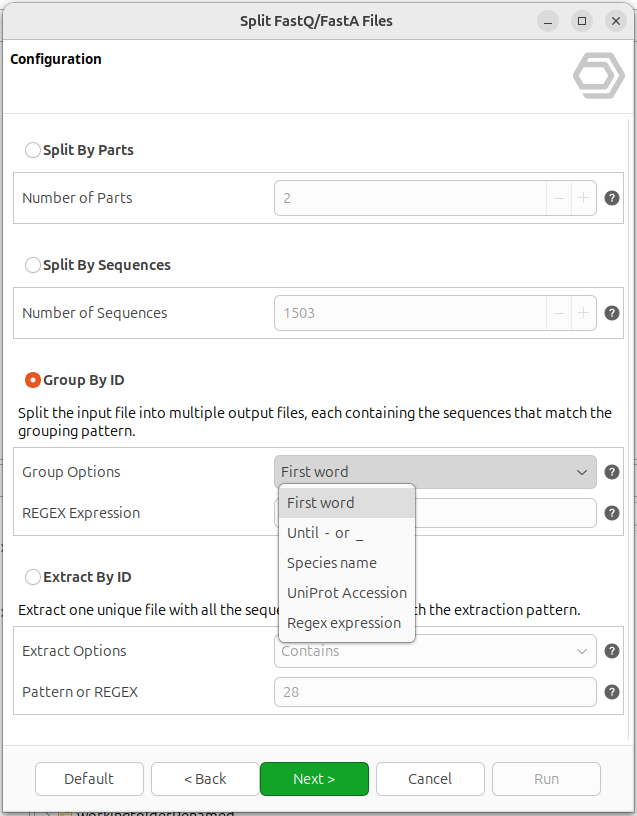
The Group By ID mode relies on regular expressions to decide how sequences should be distributed into files. To make this accessible to users with no regex experience, OmicsBox provides four predefined grouping patterns that cover the most common use cases (Figure 3):
- First word: groups sequences sharing the same first word of the ID (everything up to the first whitespace).
- Until
-or_: groups sequences sharing the same leading characters, up to the first dash or underscore. - Species name: groups sequences by
Genus_speciesprefixes, which is a common convention in comparative genomics datasets. - UniProt Accession: groups sequences by UniProt accession, regardless of its position in the header.
A custom Regex expression option is also available for advanced users who need finer control. As a rule of thumb, very specific patterns (e.g. matching a single known ID) are better suited for the Extract By ID mode, since every unique match in Group By ID generates a new output file.
Extracting specific sequences
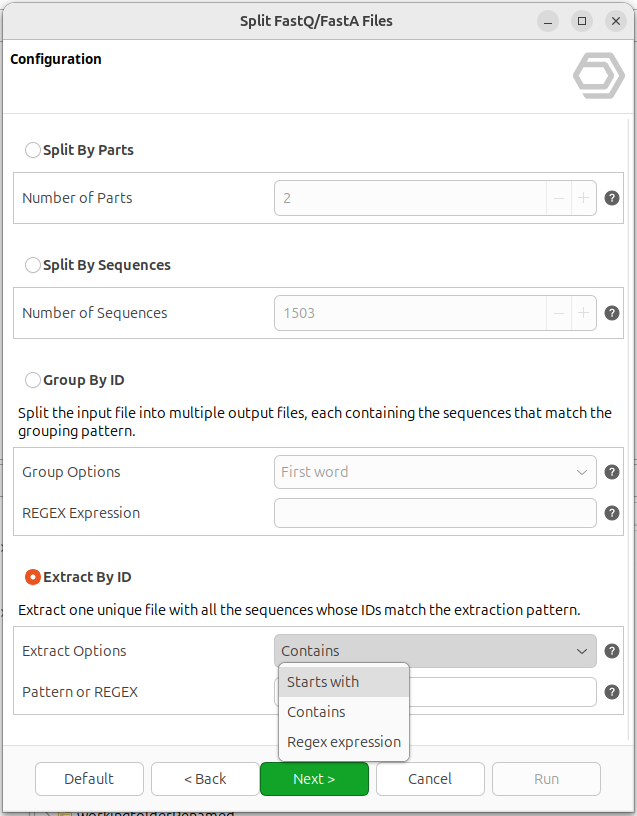
The Extract By ID mode is designed for the opposite use case: instead of partitioning the whole file, it returns a single output containing only the sequences of interest. Users can choose between three patterns (Figure 4):
- Starts with: extracts sequences whose ID starts with a given text.
- Contains: extracts sequences whose ID contains the given text in any position.
- Regex expression: full flexibility for advanced use cases.
Output
The resulting files are saved in the directory specified by the user. They are automatically named using the input file name plus a distinctive suffix, so the provenance of each output is always clear at a glance.
When splitting by parts or by number of sequences, a numeric suffix is added, for example
SRR6312174.part_001.fastq.gz or SRR6312174.part_002.fastq.gz.
When grouping by ID, the matched ID pattern is used directly as the suffix for each file, for example hairpin.part_hsa.fa or hairpin.part_mmu.fa
Key Points
- Split By Parts: create equally-sized chunks for parallel processing.
- Split By Sequences: respect per-file sequence limits in downstream tools.
- Group By ID: separate multi-species or multi-source FASTA files into biologically meaningful subsets.
- Extract By ID: pull a specific subset of sequences out of a large database without writing a single line of code.
Conclusion
The new Split FASTQ/FASTA Files tool in OmicsBox 4.0 brings one of the most common preprocessing tasks, traditionally tied to the command line, into a fully graphical, user-friendly workflow. By combining the performance of SeqKit with the intuitive OmicsBox wizards, users can now prepare their sequence files for any downstream analysis without scripting, regex headaches, or memory issues.
Because the tool is integrated in the same environment as the rest of OmicsBox, the output files can be immediately used as input for any other analysis available in the platform, from quality control and genome assembly to functional annotation, variant calling, or transcriptomic workflows. This makes OmicsBox an end-to-end solution for modern bioinformatics, from raw data to biological insight.
To learn more about this and other tools available in OmicsBox, visit the OmicsBox User Manual or explore the full list of features at OmicsBox by BioBam.
References
Shen W., Le S., Li Y., Hu F. (2016). SeqKit: A Cross-Platform and Ultrafast Toolkit for FASTA/Q File Manipulation. PLoS One, 11(10), e0163962. doi: 10.1371/journal.pone.0163962
OmicsBox – Bioinformatics Made Easy, BioBam Bioinformatics, OmicsBox by BioBam.
About the Author
 Guillem Arbiol
Guillem ArbiolGuillem is a computer engineer and holds a Master’s in Bioinformatics and Computational Biology from the Autonomous University of Madrid. He combines a strong background in software development with experience in bioinformatics data analysis, contributing to multidisciplinary projects. Guillem is currently part of the core development team behind OmicsBox.

