The Burrows-Wheeler Alignment algorithm (BWA) is a tool designed to align short DNA and RNA reads. BWA is based on the Burrows-Wheeler Transform (BWT), an algorithm designed to rearrange sequences into runs of similar characters to improve data compression and efficient sequence alignment in Bioinformatics. BWA supports both base space reads, e.g. from Illumina sequencing machines, and color space reads from AB SOLiD machines.
Whether you are exploring genetic variations, structural variants, or population genetics, aligning short reads is the first step that needs to be executed. In addition to this DNA-alignment purposes, BWA can also be used for RNA alignment, which is especially useful in prokaryotes, as they do not have splicing events. Using BWA within OmicsBox streamlines the alignment process, offering a user-friendly experience. With a practical interface, OmicsBox will guide you through the alignment process.
Using BWA in OmicsBox
Setting Up BWA in OmicsBox
BWA can be found inside the Genome Analysis and the Transcriptomics modules. Once BWA is selected, you will have to enter your input reads, and the reference genome, and you will be able to tweak different parameters. To do so, three different wizard pages will appear:
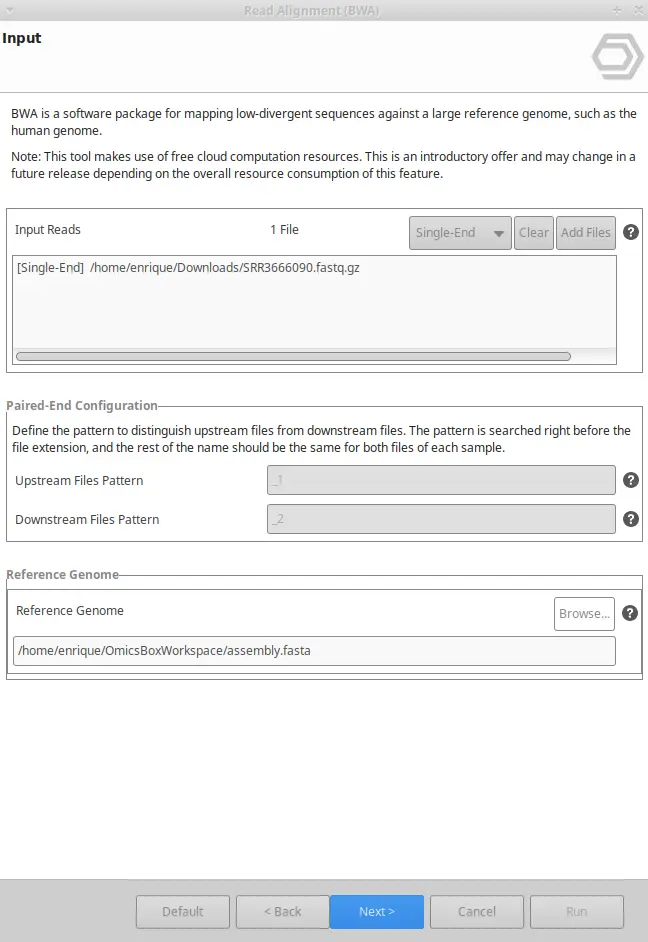
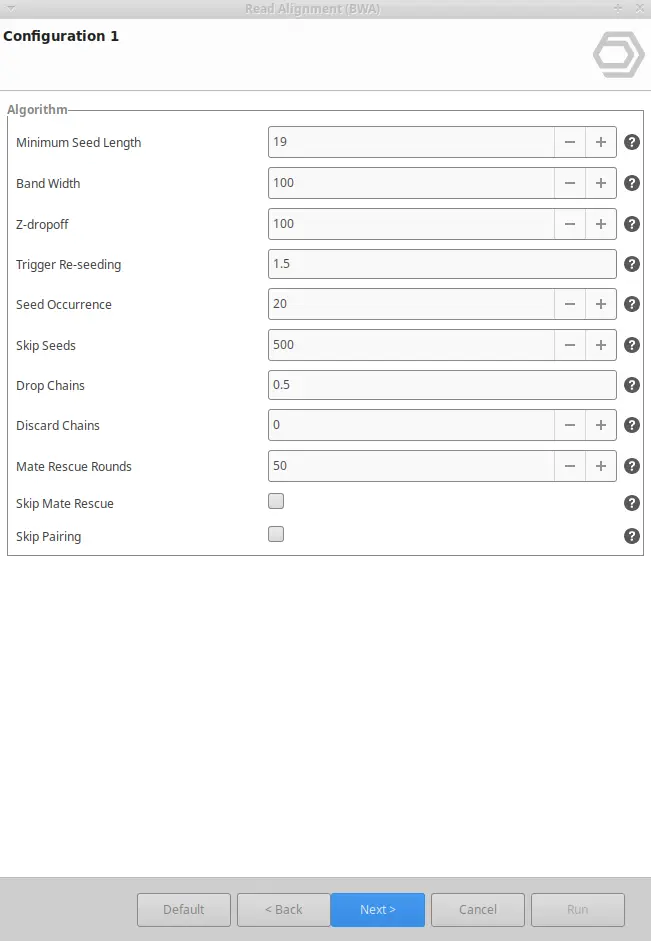
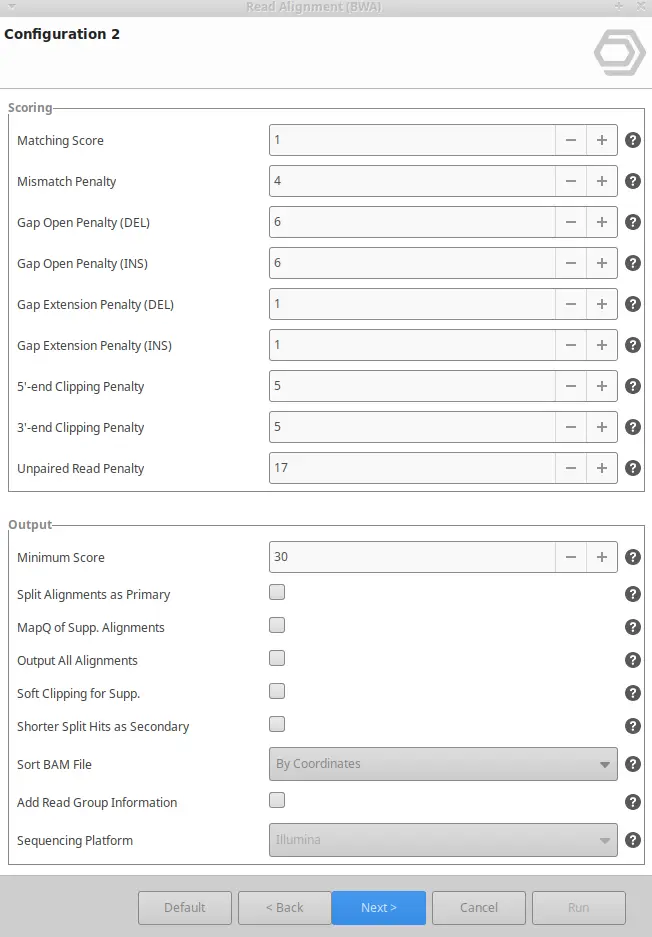
BWA output in OmicsBox
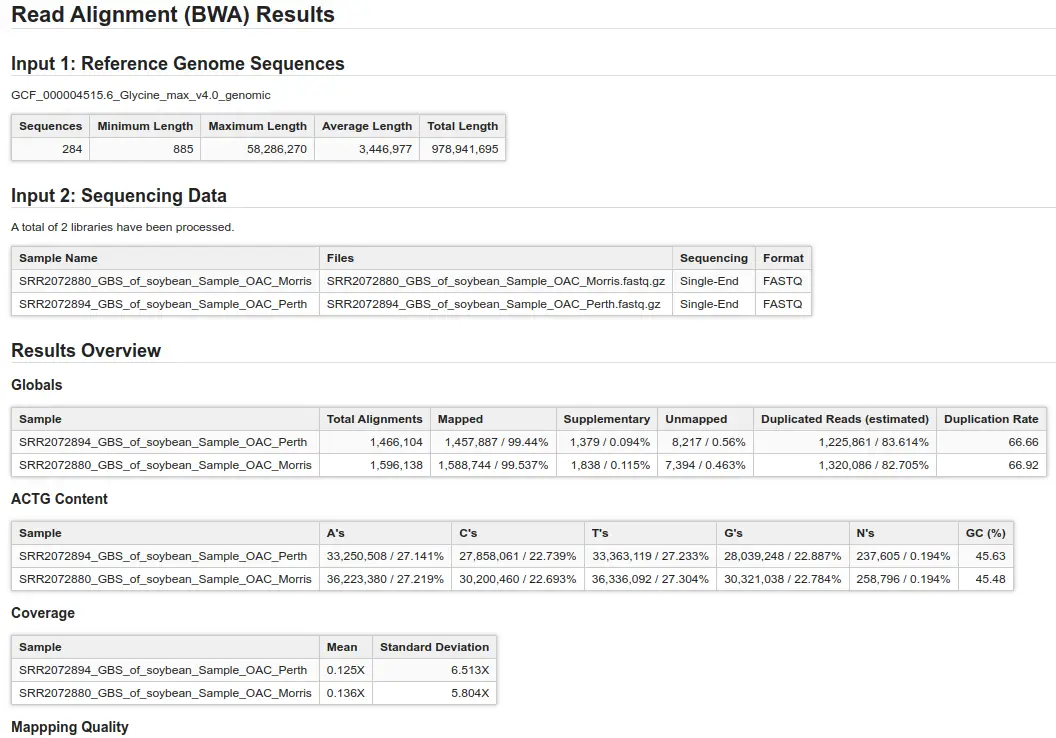
Once BWA is finished, you will receive one BAM file per sample used as input. Nevertheless, OmicsBox will also provide a summary report and charts to assist in evaluating the alignment of your files:
- Summary Report: This detailed report provides insights into the quality of your alignment. It includes information on alignment coverage, mapping quality, and other metrics to help determine the optimization of your input files and parameters.
- Alignments per Category: This section displays the number of aligned and unaligned reads, presented in both absolute and relative values.
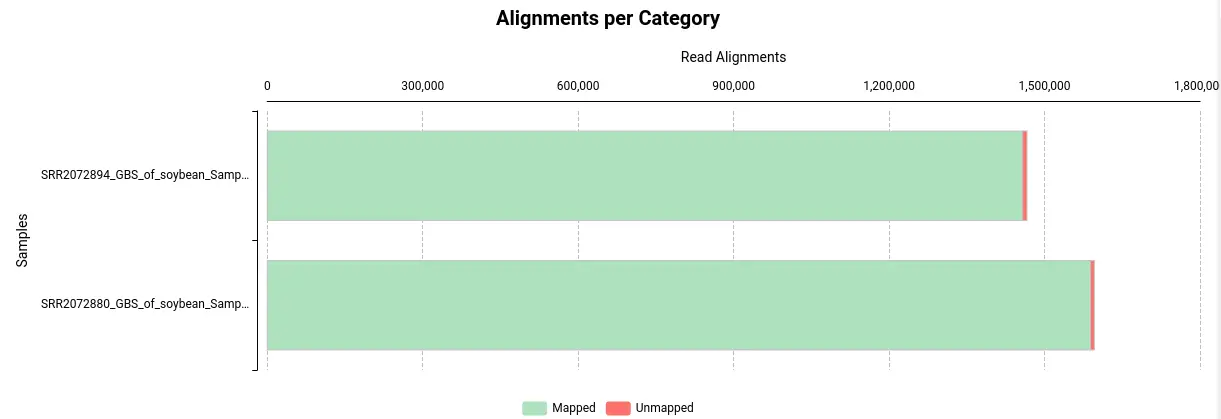
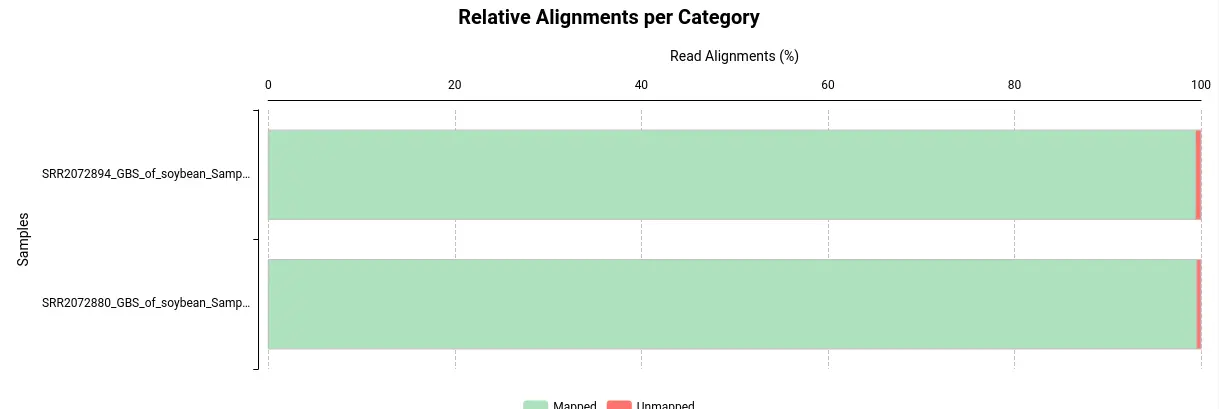
Exploring BAM files with the Genome Browser
Once you have obtained the BAM files with your alignments, you will be able to load them onto our Genome Browser to see the alignments all over the genome. This is particularly useful when you are interested in a particular region of the genome and you want to see which samples have reads aligned there and what is the sequencing depth. In addition, the genome browser will also highlight the differences between the read and the reference sequence.

Conclusions
- The alignment of DNA and RNA short reads serves as the initial step for various analyses, such as Variant Calling, Structural Variation Analysis, or Differential Expression Analysis.
- OmicsBox is a useful tool to run standard bioinformatics analysis in a user-friendly environment and obtain additional information.
- The integration of BWA within OmicsBox presents a significant opportunity to align data and then continue with downstream analysis and see your aligned reads in the Genome Browser.
References
Li, H., & Durbin, R. (2009). Fast and accurate short read alignment with Burrows–Wheeler transform. bioinformatics, 25(14), 1754-1760.
OmicsBox – Bioinformatics Made Easy, BioBam Bioinformatics, March 3, 2019, OmicsBox by BioBam
About the Author

With a biological and technological academic background, including a BSc in Biotechnology and an MSc in Bioinformatics, Enrique’s expertise lies in the areas of Long Reads and Genetic Variation.

