The study of genetic variation is fundamental in genomics research as it enables the comprehension of mutation causes and effects in the diversity of life. However, research in this field is well-developed in model organisms, while non-model organisms lack sufficient guidance for a consistent variant analysis pipeline. OmicsBox is a robust tool for investigating genetic variation in non-model organisms, facilitating the transition from variant discovery to comprehensive analysis. This blog provides a clear and concise introduction to effectively analyzing genetic variants in non-model organisms using OmicsBox.
Genetic Variation Analysis in OmicsBox
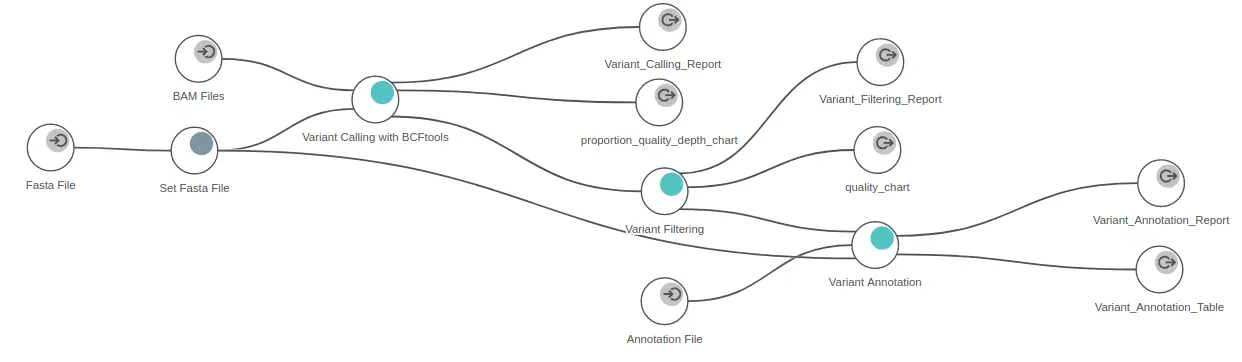
Analyzing variation in OmicsBox is straightforward. To annotate variants in non-model organisms, OmicsBox is suitable as it only requires files containing aligned sequences, a reference genome, and an annotation file. This is particularly useful since non-model organisms often have limited available information.
Step 1: Variant Calling
The first step involves discovering the variants present in the alignment files. OmicsBox offers two variant callers for non-model organisms: BCFtools and Freebayes. The former is recommended for diploid organisms, while the latter is suitable for polyploid species. However, whichever algorithm is chosen, well-explained wizards will guide you through all the settings necessary to run the variant calling step.
Upon running the variant calling in OmicsBox, users obtain a VCF file that contains information about the discovered variants, a summary report, and several charts that show the distribution of different quality parameters (Fig. 1). With this information, users can choose the appropriate parameters to filter out unreliable variants and only retain the ones that meet their standards for further analysis.
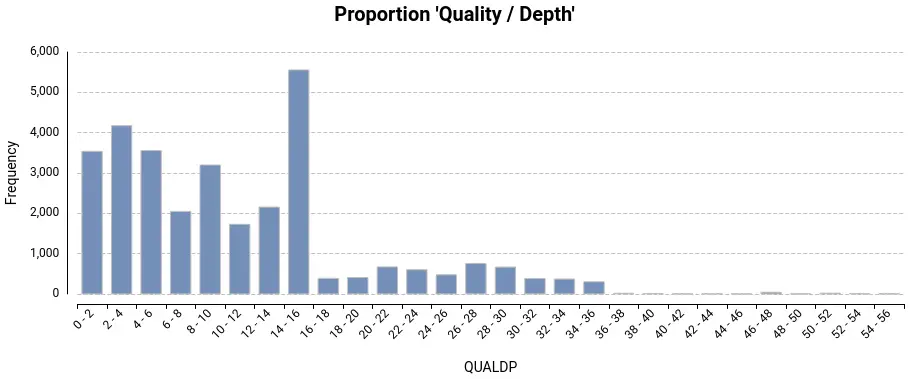
Step 2: Variant Filtering
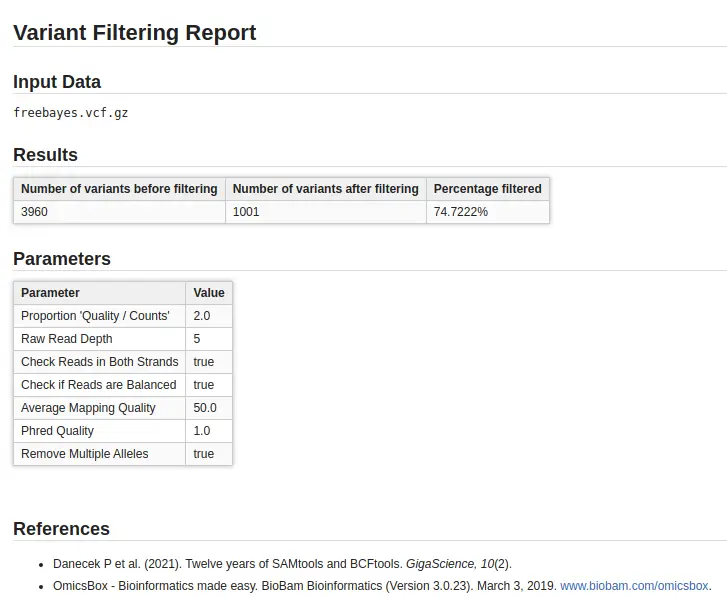
The filtering analysis identifies highly confident variants and removes falsely called ones. Although no unique canonical criteria exist, OmicsBox provides a variant filtering tool that gathers the main parameters to filter both BCFtools and Freebayes VCF files. Additionally, distribution charts of quality parameters help set different thresholds and filter out low-quality variants easily.
Following this, users receive a filtered VCF file, a report displaying the number of retained variants (Fig. 2), and updated distribution charts. Based on this information, users can decide whether to continue with the analysis or adjust the thresholds for the filtering step.
Step 3: Variant Annotation
After obtaining a VCF file with highly confident variants, OmicsBox can quickly annotate them. This requires a reference genome in FASTA format and an annotation file in GTF or GFF format, making it a suitable tool for annotating variants in non-model organisms. It’s important to note that the same reference genome must be used for both the alignment and the annotation steps.
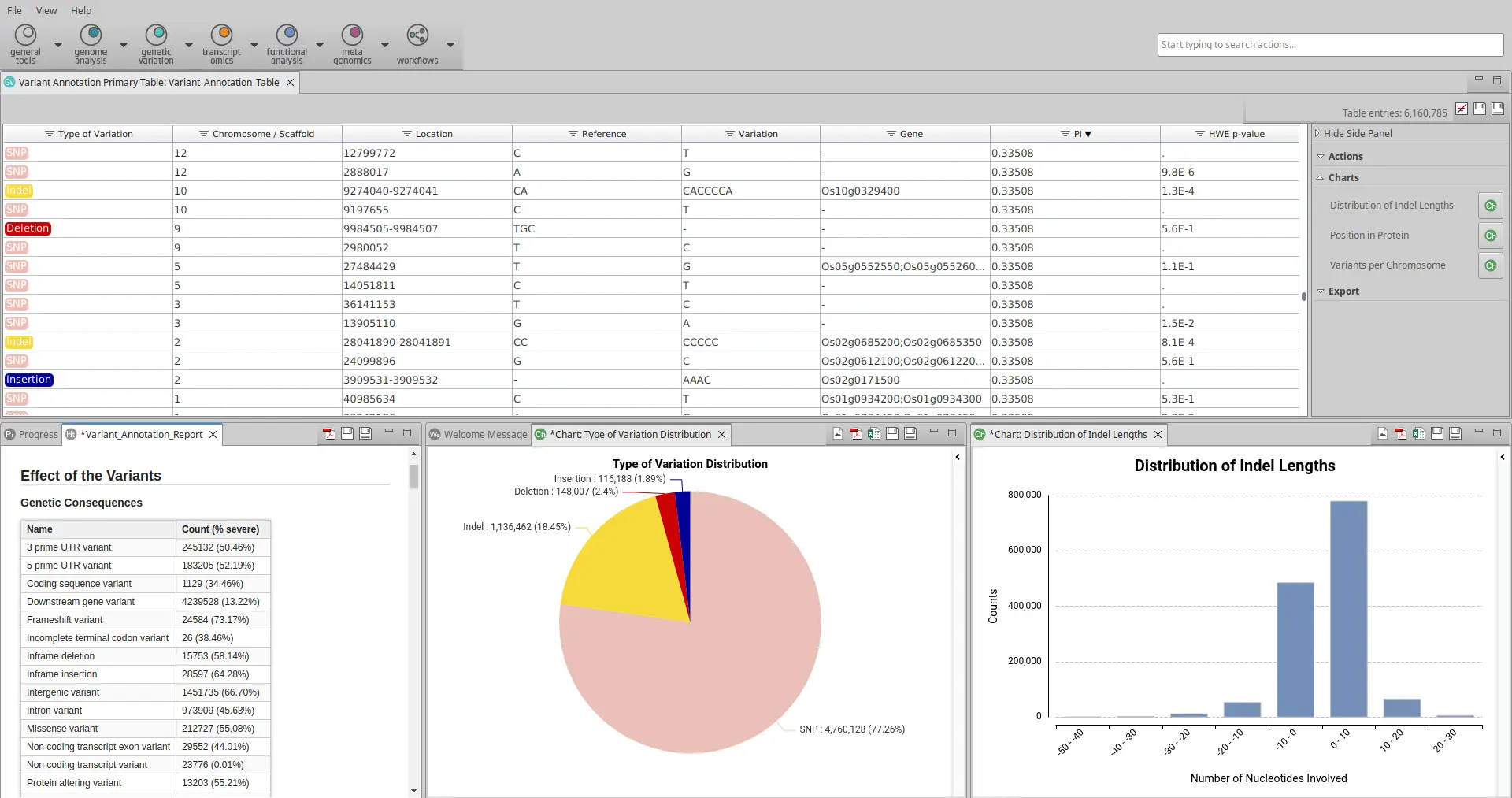
The Variant Annotation tool in OmicsBox provides comprehensive information as:
- A table with the position of each variant, the genes that are affected by them, and two population genetics values: Pi and the Hardy-Weinberg equilibrium p-value (Fig. 4).
- A summary report with information on the genetic and coding consequences and the heterozygosity of samples.
- A pie chart with the distribution of the types of variants.
- Quality control charts.
To determine precisely how a variant affects a gene, users can access the ‘Show Annotation Details’ option by right-clicking on the gene of interest to identify which transcripts or other features are affected.
Genetic Variation in OmicsBox
- OmicsBox requires only three files – aligned sequence files, a reference genome, and an annotation file – to perform genetic variation analysis. This streamlined process makes it easy to perform genetic variation analysis in all kinds of situations.
- OmicsBox is suitable for annotating variants in non-model organisms. This is particularly important because non-model organisms often lack the genetic information available for model organisms, making it difficult to perform genetic variation analysis.
- OmicsBox offers well-explained wizards that guide users through all the necessary steps to run the variant calling and filtering steps. Additionally, OmicsBox provides several quality control charts and summary reports that allow users to filter out unreliable variants and select only the most confident variants for further analysis.

References
Danecek, P., Bonfield, J. K., Liddle, J., Marshall, J., Ohan, V., Pollard, M. O., … & Li, H. (2021). Twelve years of SAMtools and BCFtools. Gigascience, 10(2), giab008.
McLaren, W., Gil, L., Hunt, S. E., Riat, H. S., Ritchie, G. R., Thormann, A., … & Cunningham, F. (2016). The Ensembl variant effect predictor. Genome biology, 17(1), 1-14.
Useful links
About the Author
 Enrique Presa
With a biological and technological academic background, including a BSc in Biotechnology and an MSc in Bioinformatics, Enrique's expertise lies in the areas of Long Reads and Genetic Variation.
Enrique Presa
With a biological and technological academic background, including a BSc in Biotechnology and an MSc in Bioinformatics, Enrique's expertise lies in the areas of Long Reads and Genetic Variation. 
