In this analysis, we will reproduce a study carried out by Michaela M. Martis et al. in 2017 with OmicsBox.
(http://doi.org/10.1371/journal.pone.0185182)
Introduction
Ascaridia galli is an intestinal parasite that infects a wide range of domestic birds. It is especially important in European farms, where it parasites laying hens and causes economic problems. The only available treatments are the Benzimidazole derivatives, such as Flubendazole (FLBZ), a group of anthelmintic drugs. However, their overuse could be problematic in the future, as A. galli could develop resistance mechanisms.
The objectives of this transcriptomics study were:
- To develop a reference transcriptome of A. galli.
- To understand the response in gene expression before and after the exposure to FLBZ through an RNA-Seq Differential Expression Analysis.
- To investigate b-tubulin transcripts expression and phylogeny, several studies have reported the relationship between b-tubulin and Benzimidazole resistance.
1. Data Download and Preprocessing
Firstly, we downloaded raw paired-end sequenced reads from SRA through the fastq-dump tool, from SRA-Toolkit. We obtained six fastq files, one per analyzed sample, and we classified them according to the experimental design, which is described below. Our complete dataset contained about 173 million reads.
We used Trimmomatic in order to remove the Illumina adapters. Trimmomatic is command-line software that removes the adapter sequences from the sequenced raw reads. This tool has already been incorporated into OmicsBox (General Tools > FastQ Tools > FastQ Preprocessing).
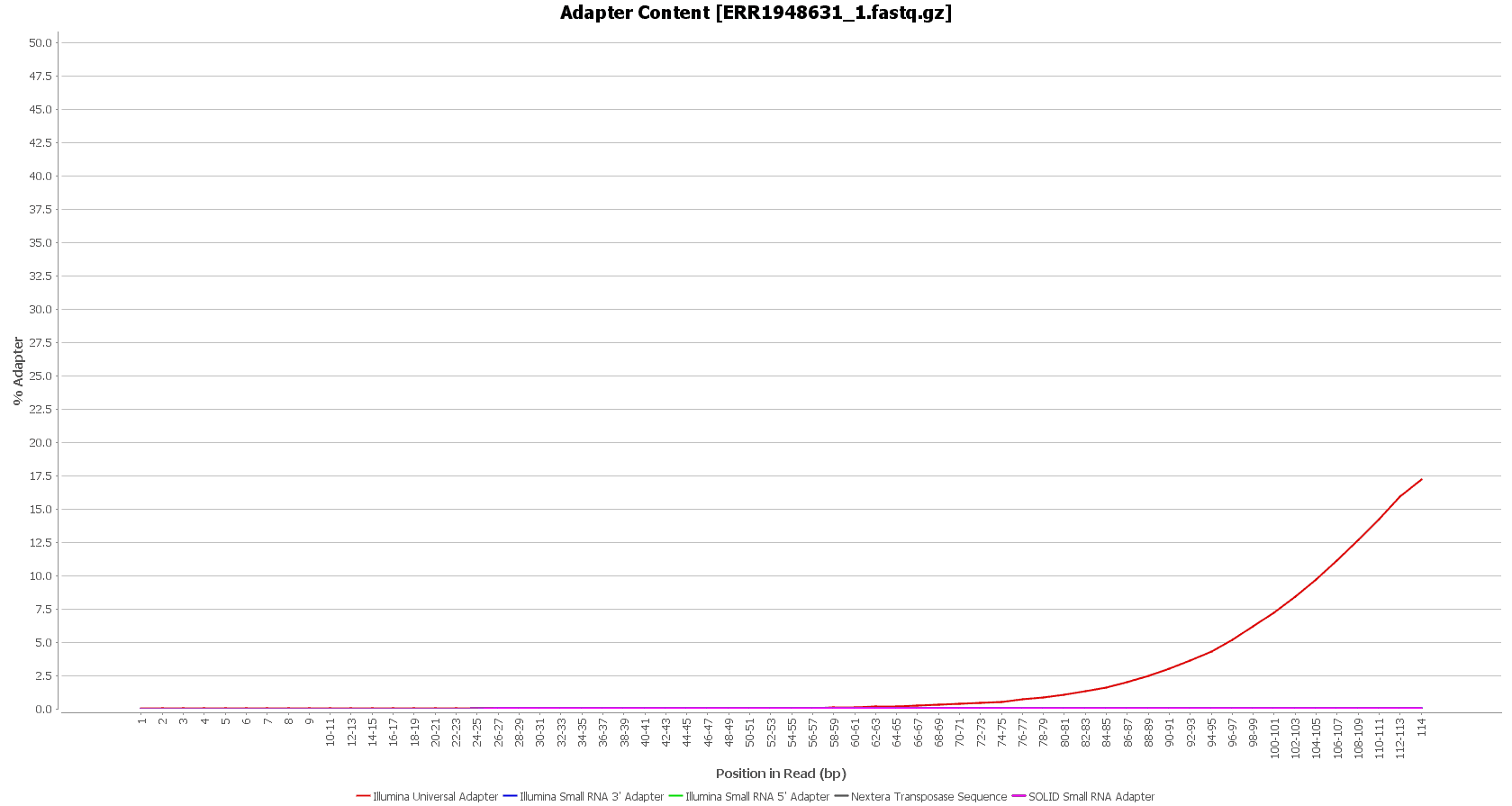
2. De novo Transcriptome Assembly and Completeness Assessment
We performed a de novo transcriptome assembly using the cleaned reads obtained from Trimmomatic. We used Trinity, in OmicsBox (Transcriptomics > Assembly > RNA-Seq de novo assembly) in the non-strand-specific mode. After this step, we obtained an assembled transcriptome with 290713 transcripts.
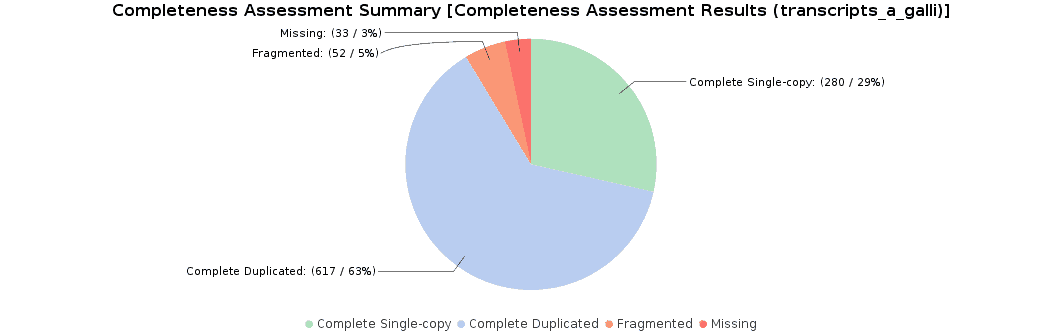
To assess the completeness of our transcriptome assembly, we searched for nematode orthologs with BUSCO (Benchmarking Universal Single-Copy Orthologs) with OmicsBox. This feature can be found under Transcriptomics > Assembly > Completeness Assessment. In the light of the results obtained, we concluded that we had generated a good quality transcriptome: 92% of nematode orthologs have, at least, single-copy representation in our assembly, while 5% of them were partially found and 3% were missing. Furthermore, we achieved similar findings to those resulting from the original study (they found 91.9% complete orthologs, and 3.3% were missing).
3. Open Reading Frame Prediction
Once we obtained our transcriptome, we searched for ORFs within the predicted transcripts with OmicsBox (Transcriptomics > Assembly > Predict Coding Regions). This tool extracts and classifies the best ORFs from a transcripts dataset on the basis of Pfam domain search data.
90000 ORFs were predicted and classified according to their completeness: 73.3% of transcripts were complete, 16.6% with a missing start codon, 6.1% with a missing stop codon, and 4.0% lacked both start and stop codons. These results were pretty similar to the original ones, where 119000 ORFs were predicted and classified.
![]()
After finding the Open Reading Frames, we had to filter this output in order to keep the best ORF per gene. As A. galli is a simple organism, we assumed that actually there was only one isoform per gene. This “best” isoform was selected by keeping the longest one for each gene. After that, 19700 genes were kept for further analysis.
4. Functional Annotation and InterProScan
We used the Blast2GO methodology (Functional Analysis Module) to perform a functional annotation with default parameters and an InterProScan analysis of the TransDecoder predicted coding transcripts, as they were our new “coding transcriptome”. We followed these steps:
- BlastX against SwissProt database.
- CloudIPS against HMMPIR, HAMAP, and HMMPanther, and IPS against TMHMM.
- Mapping and annotation.
- Merge IPS results to annotation.
Once the annotation step was finished, we obtained a functional annotation for 8,661 genes, which means that 44% of the TransDecoder transcripts had a functional annotation.
5. Coding Transcripts Filtering and Classification
In addition, our 19,700 transcripts were classified into “high confidence (HC)”, if they had Blast results and an ORF predicted; and “low confidence (LC)”, if they only met one criterion. 85% of genes were classified as “high confidence”, and the remaining ones as “low confidence”.
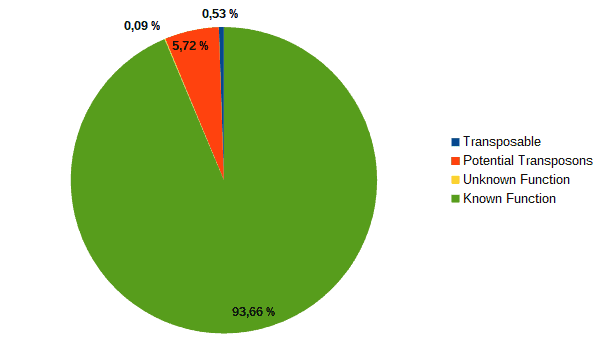
Then, according to the article, we classified transcripts from both datasets into four groups according to their function: “genes with an assigned function”, “genes with unknown function”, “transposons” or “potential transposons” by examining their description in OmicsBox. Most of the LC genes had an unknown function or acted as transposable elements, while the HC ones were classified as genes with an assigned function.
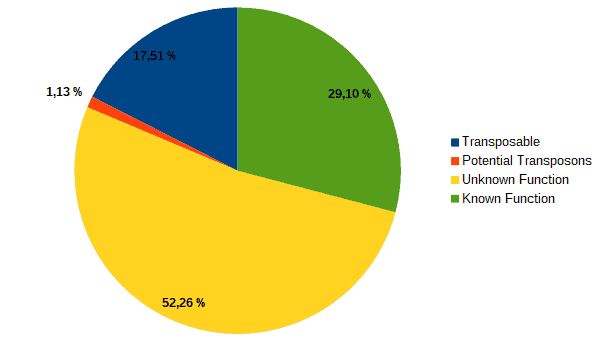
6. Differential Expression Analysis (DEA)
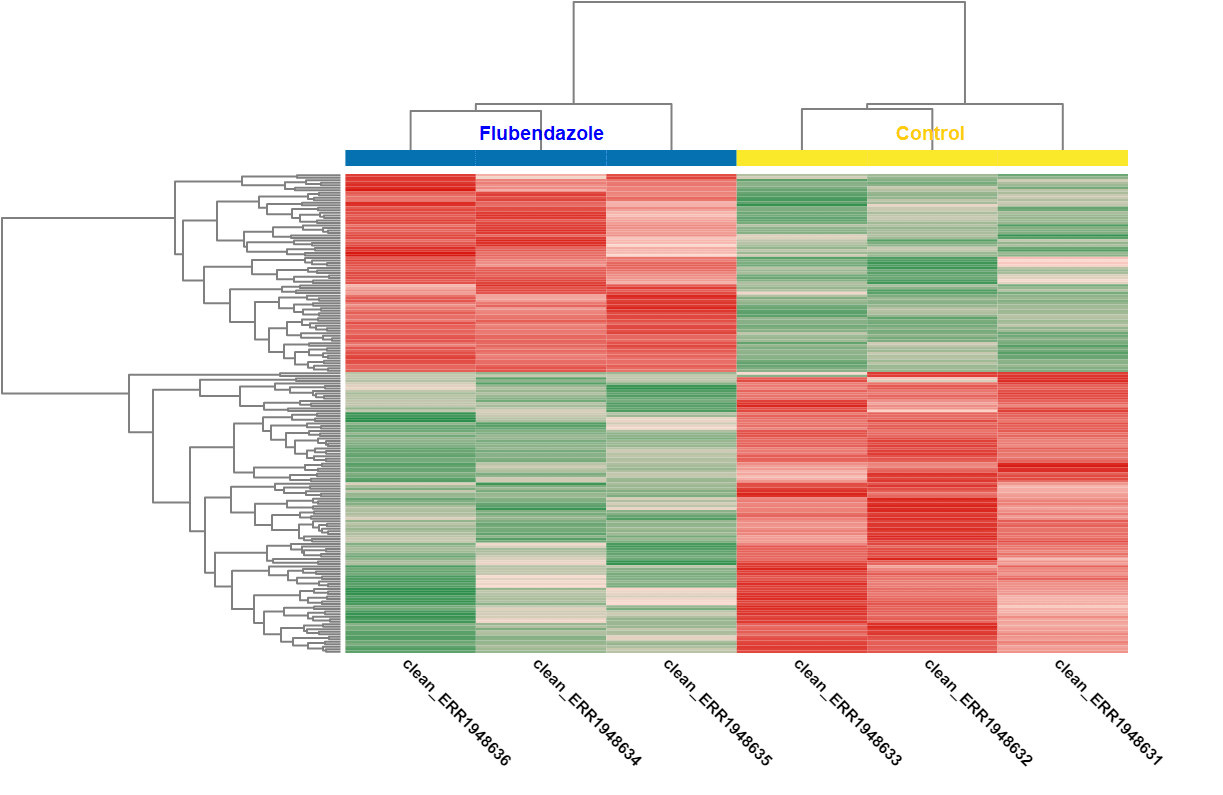
A DEA was carried out in OmicsBox. First, we created a count table (Transcriptomics > Create Count Table > Transcript-level Quantification) with our filtered coding transcriptome and the clean reads we generated in step 1. Then, we performed a DEA (Transcriptomics > Pairwise Differential Expression Analysis) with stringent criteria, and we used the experimental design described above to visualize the differentially expressed genes. We detected 58 up- and 89 down-regulated genes, while the authors found 110 up- and 153 down-regulated ones.
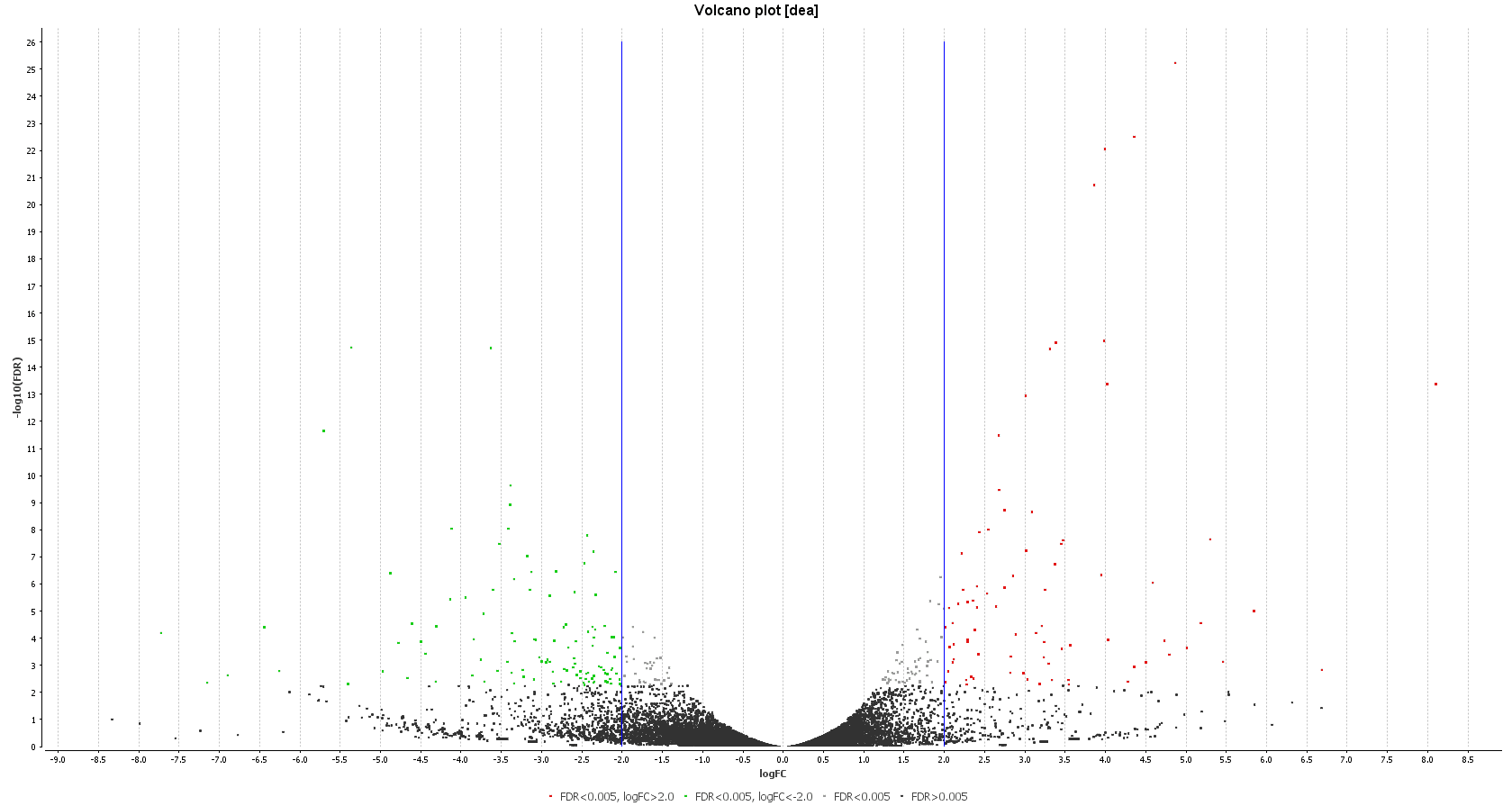
Then, we searched for up-regulated genes related to stress or drug resistance by using the OmicsBox description filter, and we obtained similar results to those reported by the authors. We found two candidates:
- Multidrug resistance protein (PGP). A member of the ATP-binding cassette superfamily. The resistance is associated with a decrease in drug accumulation and an increase in drug efflux.
- Heat shock protein (HSP). A subset of a group of proteins produced in response to exposure to a range of stressful conditions.
Another gene we found was phosphofructokinase (PFK), an enzyme that serves as a key regulatory step in the glycolysis pathway. This finding suggests that glycolysis could be affected by exposure to FLBZ.
Within down-regulated genes, we found:
- Glutamate dehydrogenase (GDH). It is an enzyme with a key role in energy homeostasis in nematodes, with reactions involved in ATP production.
- Cytochrome P450. It is involved in ATP production via oxidative phosphorylation.
Both findings suggest that ATP production is affected by FLBZ.
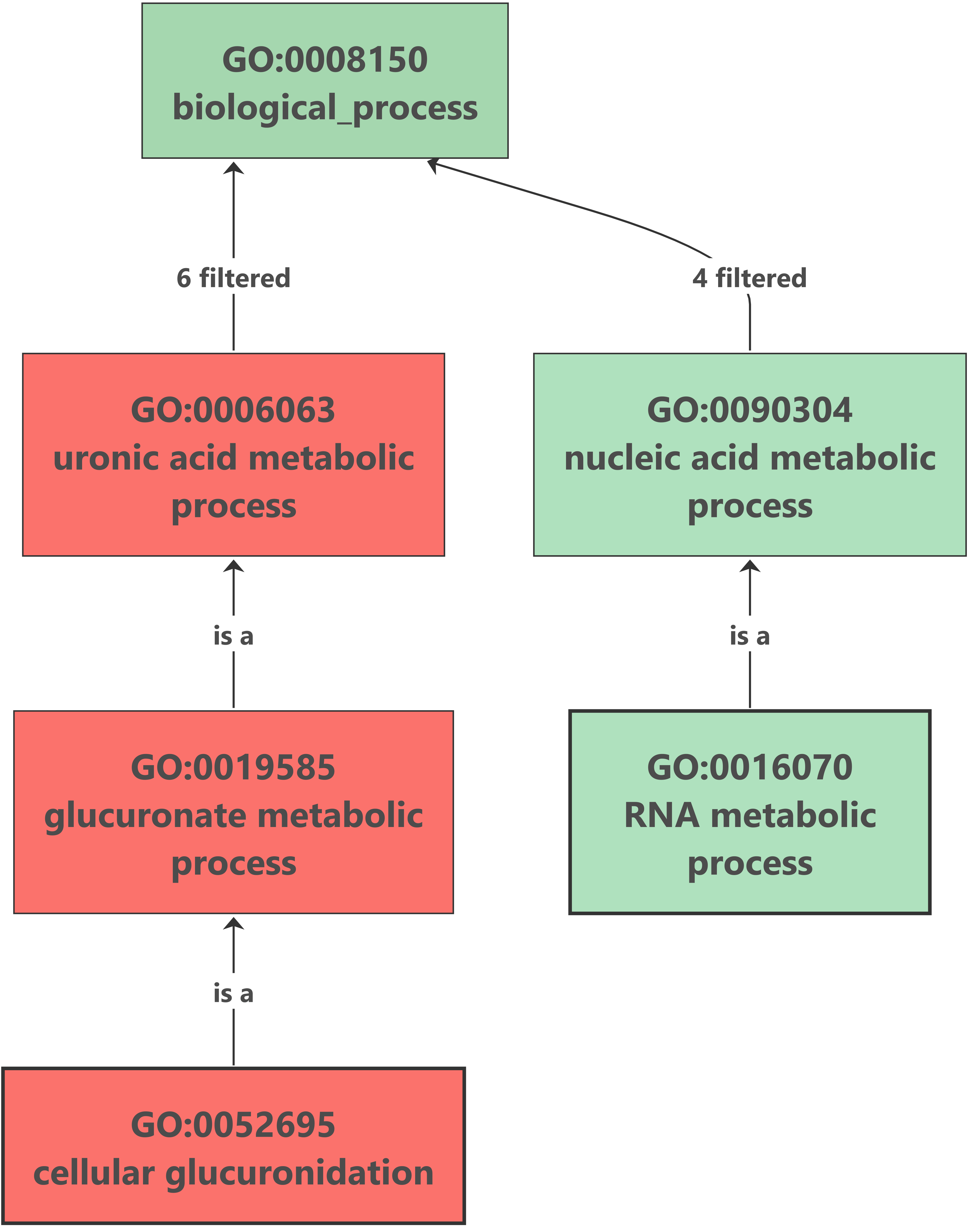
7. Enrichment Analysis – Fisher’s Exact Test
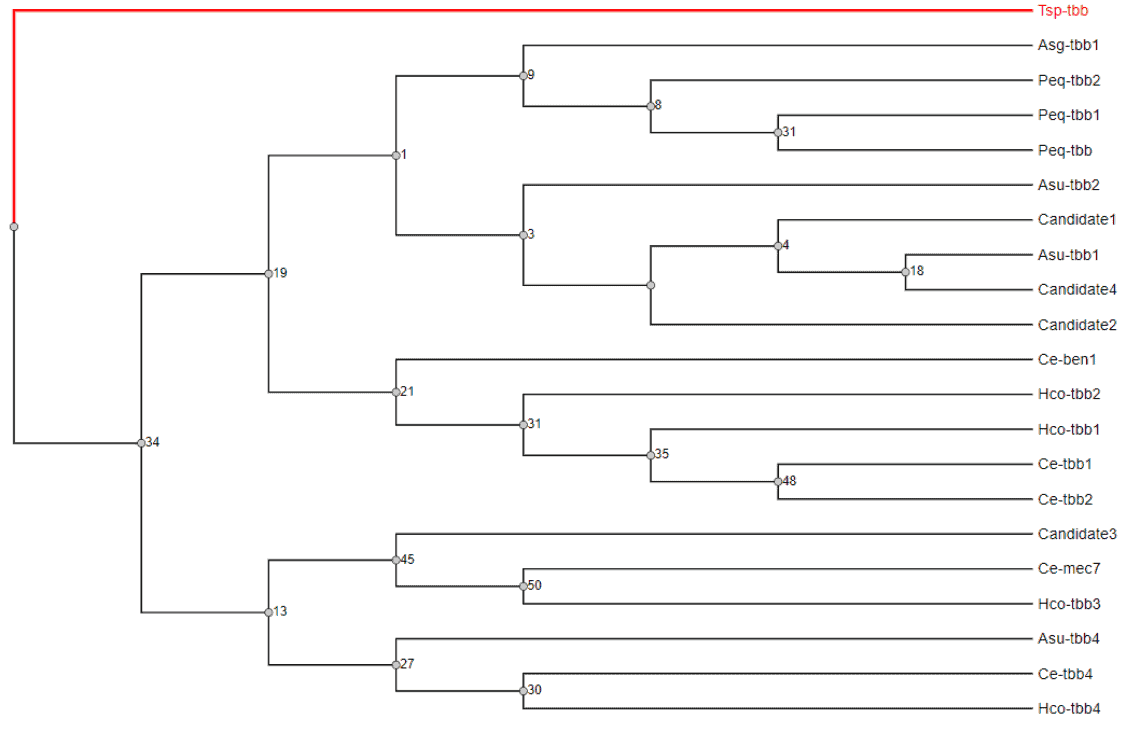
8. Candidate Transcripts Tesponsible for Anthelmintic Resistance: b-tubulin Phylogenetic Analysis
B-tubulins are cytoskeletal proteins that are suspected to be related to benzimidazole resistance, as these drugs seem to exert selection pressure in some b-tubulin isoforms. We performed a phylogeny in order to know how identified b-tubulin transcripts from A. galli were related to other tubulins from closely related species.
We downloaded some b-tubulin sequences from C. elegans, A. suum, A. galli, P. equorum and H. contortus as related species. In addition, we obtained a b-tubulin sequence from a remote species, T. spiralis, which was used as an outgroup sequence, to root the tree. Then, we identified four b-tubulin candidates in our transcriptome by using the OmicsBox filters (but none of them was up or down-regulated).
Conclusions
- Our de novo transcriptome assembly and the differential expression analysis provided some information about the A. galli response to flubendazole.
- We used BUSCO, a software which offers information about the assembly quality and completeness. It is an easy-to-use tool and provides a good method to validate an assembly.
- OmicsBox allowed us to assemble the A. galli transcriptome, annotate it, identify the description of up and down-regulated genes, and perform enrichment analysis.
- We identified up-regulated genes associated with stress conditions, drug excretion, and with the glycolysis pathway.
- Down-regulated genes were associated with ATP and energy production.
- Contrary to what was expected, our enrichment analysis showed that glucuronate metabolism, a drug elimination pathway, is overrepresented among down-regulated genes.
- Our phylogenetic analysis of b-tubulin sequences, which was carried out with MAFFT and PhyML, suggested that these proteins might not be involved in the development of drug resistance.
References
- Martis MM, Tarbiat B, Tydén E, Jansson DS, Höglund J (2017) RNA-Seq de novo assembly and differential transcriptome analysis of the nematode Ascaridia galli in relation to in vivo exposure to flubendazole. PLoS ONE 12(11): e0185182.
- Götz S, et al. (2008) High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acid Research, Vol. 36, pp. 3420-3435.
- Andrews S (2010) FastQC: a quality control tool for high throughput sequence data.
- Bolger AM, Lohse M and Usadel B (2014) Trimmomatic. A flexible trimmer for Illumina Sequence data. Bioinformatics, btu170.
- Bryant DM, Johnson K, DiTommaso T, Tickle T, Couger MB, Payzin-Dogru D, Lee TJ, Leigh ND, Kuo TH, Davis FG, Bateman J, Bryant S, Guzikowski AR, Tsai SL, Coyne S, Ye WW, Freeman RM Jr, Peshkin L, Tabin CJ, Regev A, Haas BJ, Whited JL. A Tissue-Mapped Axolotl De Novo Transcriptome Enables Identification of Limb Regeneration Factors. Cell Rep. 2017 Jan 17;18(3):762-776. doi: 10.1016/j.celrep.2016.12.063. PubMed PMID: 28099853; PubMed Central PMCID: PMC5419050.
- Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, et al. (2011) Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nature Biotechnology, 29(7):644-52
- Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O (2010) New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Systematic Biology, 59(3):307-21.
- Haas B, Papanicolaou A, et al., manuscript in prep. http://transdecoder.github.io
- Katoh, Misawa, Kuma and Miyata (2002) MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acid Research 30:3059-3066.
- Langmead B and Salzberg S (2012) Fast gapped-read alignment with Bowtie 2. Nature Methods, 9:357-359.
- Li B and Dewey CN (2011) RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics, 12:323.
- Robinson MD, McCarthy DJ and Smyth GK (2010) edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics, 26, pp.-1.
- Simao F, Waterhouse R, Ioannidis P, Kriventseva E and Zdobnov E (2015) BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics.
- Gomes, Ana Paula & Olifiers, Natalie & Maria Dos Santos, Michele & Simões, Raquel & Maldonado, Arnaldo. (2015). New records of three species of nematodes in Cerdocyon thous from the Brazilian Pantanal wetlands. Brazilian journal of veterinary parasitology: Orgao Oficial do Colegio Brasileiro de Parasitologia Veterinaria. 24. 324-330. 10.1590/S1984-29612015061.
- OmicsBox – Bioinformatics made easy. BioBam Bioinformatics (Version 1.2.4). March 3, 2019. www.biobam.com/omicsbox

