Sequence alignment is an essential element of genomics research, playing a significant role in aiding scientists in deciphering the intricacies of genetic data. When confronted with the task of aligning long reads generated by Pacific Biosciences (PacBio) or Oxford Nanopore Technologies (ONT) with precision and efficiency, scientists commonly opt for Minimap2 as their preferred tool (it is the most cited long-read aligner). Minimap2 has earned this distinction due to its proven track record of accurate and efficient long-read alignment, as demonstrated in numerous peer-reviewed studies and benchmarking analyses within the genomics community (Yildiz et al., 2023; Becht et al., 2021; Bolognini & Magi, 2021; Coster et al., 2019; Zhou et al., 2019). In this blog post, we will explain the characteristics and applications of this long-read aligner and how to utilize it advantages within OmicsBox.
Minimap2 Characteristics
Minimap2 is a high-efficiency sequence alignment tool distinguished by its balance of speed, accuracy, and versatility.
- Speed: Minimap2 is designed for efficient alignment. This becomes especially important for researchers when handling sizable datasets. Within OmicsBox, this efficiency is potentially enhanced due to the cloud-based platform’s integration.
- Accuracy: Precise alignment is pivotal for deriving meaningful genomic interpretations. Minimap2 aims to reduce false positives and false negatives, striving for consistent results. Several benchmarks (Yildiz et al., 2023; Bolognini & Magi, 2021; Coster et al., 2019; Zhou et al., 2019) recommend this aligner in order to obtain structural variants, which is a task that requires the highest accuracy.
- Versatility: Minimap2 is compatible with both PacBio and Nanopore data. It offers multiple preset options and an extensive range of parameters that can be adjusted based on the specific requirements of your dataset.
Minimap2 Applications
The alignment of long-read sequenced data is crucial for different types of bioinformatic analyses. Some of these analyses are already implemented in OmicsBox, while others may be included in the future.
- Transcriptome Assembly: Constructing complete transcriptomes from long-read data requires precise sequence alignment. There are multiple approaches for this task; some are reference-guided, and others are reference-free. Currently, OmicsBox offers FLAIR, a reference-guided transcriptome reconstructor.
- Transcript Quantification with Long Reads: To generate a long-read count matrix, it’s necessary to align reads to a reference genome. Various tools can perform this quantification, including FLAIR.
- Structural Variant Detection: Identifying structural variants associated with diseases such as cancer or genetic disorders requires a tool capable of handling complex alignments.
How to Use Minimap2 in OmicsBox
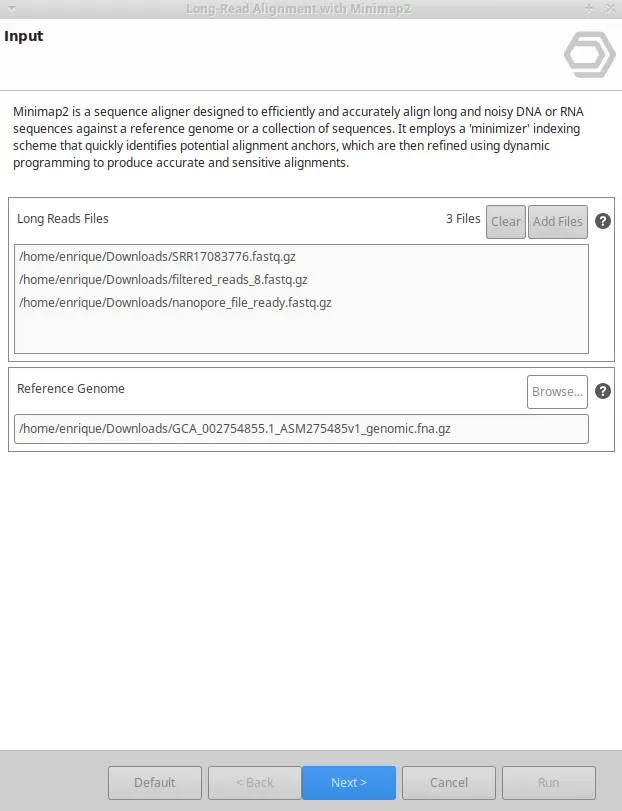
- Introduce Long-Read Data: Import the reference genome and long-read sequencing data into OmicsBox. The platform supports both FASTA and FASTQ files for maximum flexibility.
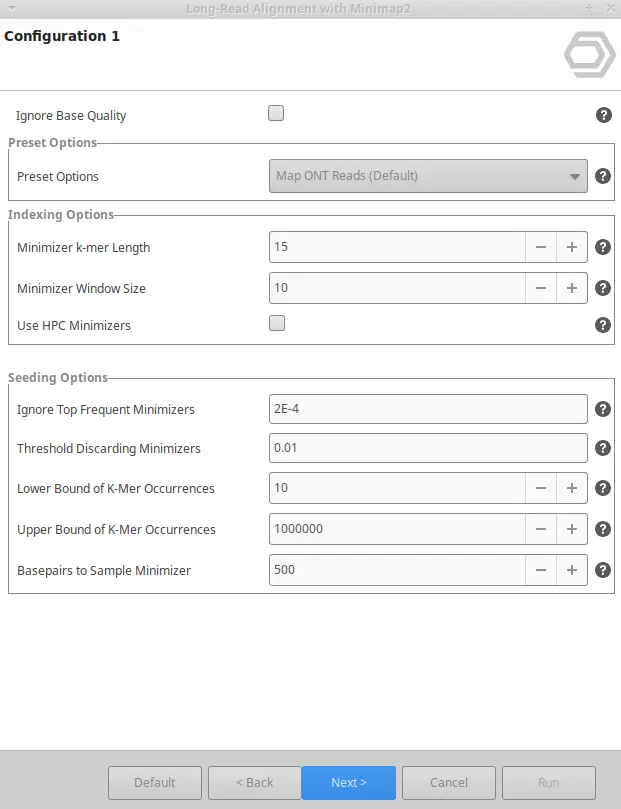
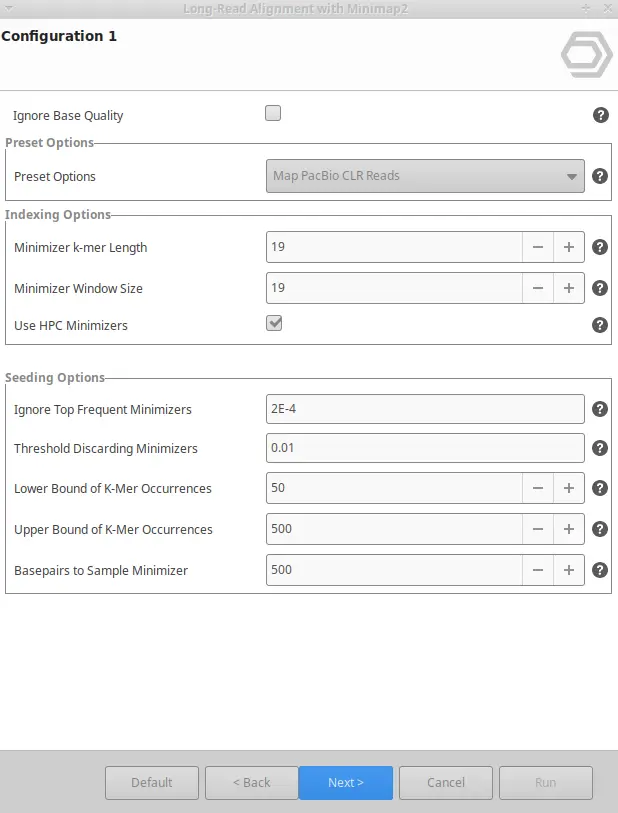
- Configure Parameters: Minimap2 has some presets of options (i.e., predefined sets of parameters) suitable for different types of data, such as PacBio HiFi reads, PacBio CLR reads, or ONT data. You can choose among different presents and, in addition, tune any other parameter in order to maximize the efficiency of your alignments.
- Run Minimap2: Once Minimap2 is running, an index of your reference file will be done, prior to the alignment of each fastQ or fastA file. You will receive one BAM file per fastQ/A file.
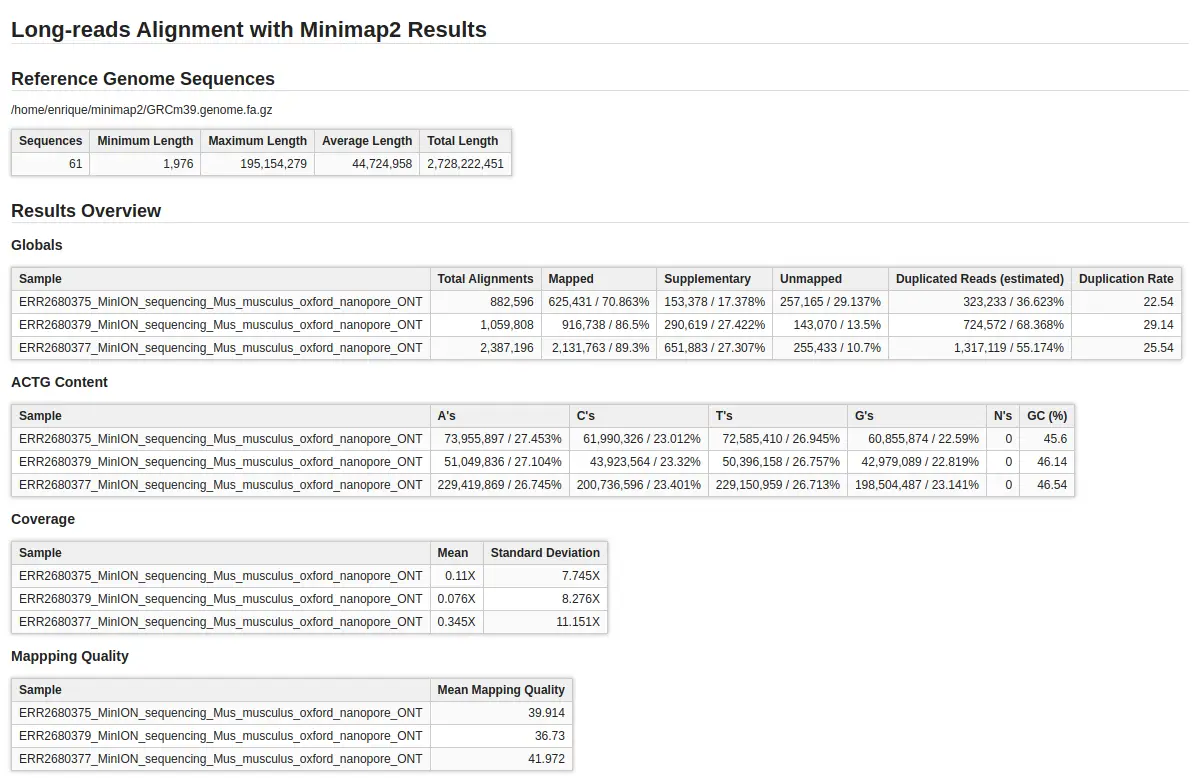
- Visualize and Analyze: Once the alignment is complete, use OmicsBox’s visualization tools and reports to explore your results. In order to do the summary report, Qualimap has been used.
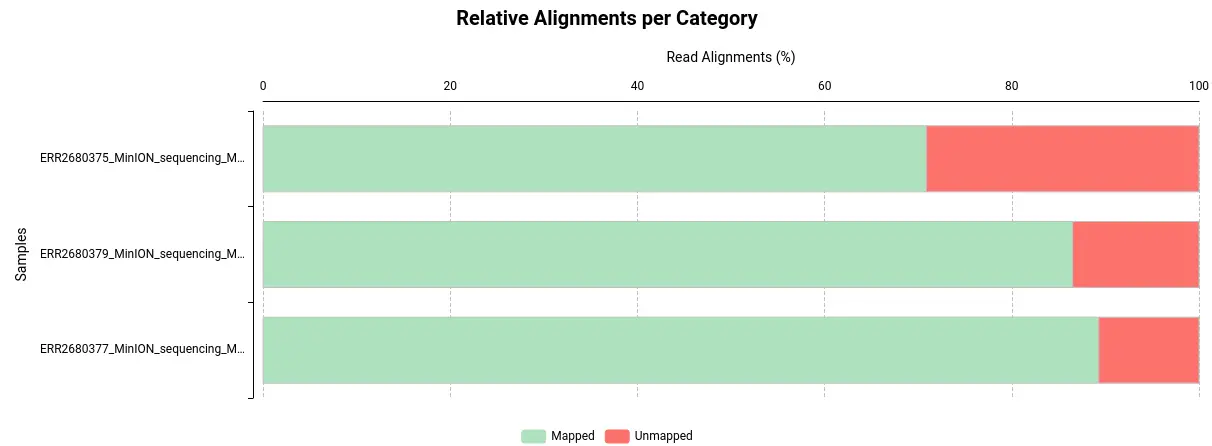
Conclusion
By running Minimap2 in OmicsBox, you will not only be able to align high volumes of long-read sequences efficiently but also streamline your entire research workflow. In addition, you will have fast results, enhanced accuracy, and a user-friendly experience that empowers researchers of all levels.
References
Yildiz, G., Zanini, S. F., Afsharyan, N. P., Obermeier, C., Snowdon, R. J., & Golicz, A. A. (2023). Benchmarking Oxford Nanopore read alignment‐based insertion and deletion detection in crop plant genomes. The Plant Genome, e20314.
Becht, C., Schmidt, J., Blessing, F., & Wenzel, F. (2021). Comparative analysis of alignment tools for application on Nanopore sequencing data. Current Directions in Biomedical Engineering, 7(2), 831-834.
Bolognini, D., & Magi, A. (2021). Evaluation of germline structural variant calling methods for nanopore sequencing data. Frontiers in Genetics, 12, 761791.
De Coster, W., De Rijk, P., De Roeck, A., De Pooter, T., D’Hert, S., Strazisar, M., … & Van Broeckhoven, C. (2019). Structural variants identified by Oxford Nanopore PromethION sequencing of the human genome. Genome research, 29(7), 1178-1187.
Zhou, A., Lin, T., & Xing, J. (2019). Evaluating nanopore sequencing data processing pipelines for structural variation identification. Genome biology, 20, 1-13.
Li, H. (2018). Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics, 34(18), 3094-3100.
Okonechnikov, K., Conesa, A., & García-Alcalde, F. (2016). Qualimap 2: advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics, 32(2), 292-294.

About the Author
 Enrique Presa
With a biological and technological academic background, including a BSc in Biotechnology and an MSc in Bioinformatics, Enrique's expertise lies in the areas of Long Reads and Genetic Variation.
Enrique Presa
With a biological and technological academic background, including a BSc in Biotechnology and an MSc in Bioinformatics, Enrique's expertise lies in the areas of Long Reads and Genetic Variation.

