Third-generation DNA sequencing technologies allows scientist to generate longer sequence reads, which can be used in whole-genome sequencing projects to yield better repeat resolution and more contiguous genome assemblies. However, although long-read sequencing technologies can produce genomes with long contiguity, the relatively high error rate of long reads has made it challenging to generate highly accurate final sequences.
OmicsBox now offers an analysis strategy to handle long-read data, such as those produced by PacBio and Oxford Nanopore technologies. It consists of three steps: assembling long reads, mapping high-quality short reads to long-read assembly, and polishing the long-read assembly with the alignments of the short reads.
In this way OmicsBox allows to assemble genomes of any size with data coming from long and short-read technologies ensuring highly contiguous assemblies with a very low overall error rate.
Long-read sequencing technologies can produce genomes with long contiguity
Long Read Assembly
Flye is a de novo assembler for single-molecule sequencing reads. It is designed for a wide range of datasets, from small bacterial projects to large mammalian-scale assemblies, and it can handle both, Oxford Nanopore and PacBio data. Flye uses the repeat graph as a core data structure. Unlike de Bruijn graphs (which require exact k-mer matches), repeat graphs are built using approximate sequence matches and can tolerate higher noise of long reads.
The Flye algorithm can be executed within OmicsBox via the DNA-Seq de novo Assembly option and its configuration is quite easy. First, provide the sequencing data in FASTQ/FASTA format. Currently, raw and corrected reads from PacBio and Oxford Nanopore Technologies are supported. Mixing different read types is not yet supported. Optionally, other assemblies, such as the ones produced by different short/long-read assemblers, can be provided to generate a consensus of multiple assemblies. Flye, to work correctly, requires as input an estimate of the genome size to be assembled in order to decide about read-correction and how sensitive the “overlapper” should be. Additionally, common Flye options can be adjusted, such as the minimum overlap and options for plasmid and haplotype detection.
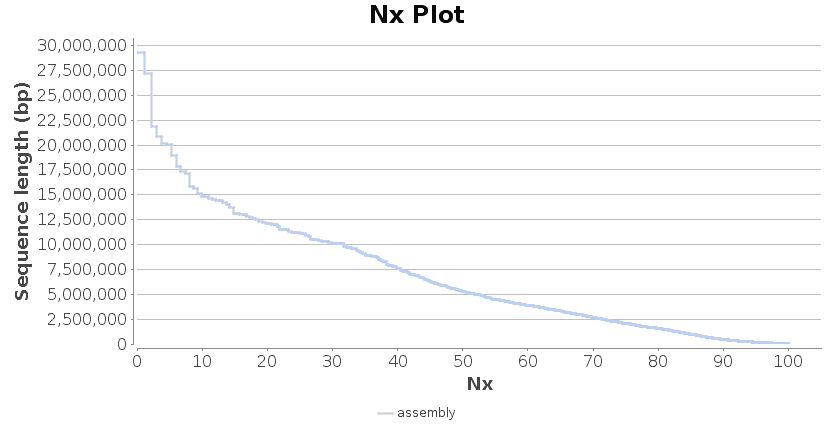
Flye returns as output a FASTA file containing the assembled sequences. Furthermore, a number of common statistics used to describe the quality of sequence assembly can be consulted in the report and the chart that are provided. The N50 statistic defines the assembly in terms of contiguity. It is calculated by first ordering every Contig from the longest to the shortest, and then, the length of each sequence are summed up until this running sum equals one-half of the total length of the assembly. The length of the shortest Contig in this list is the N50 value. Higher values of N50 indicate a better assembly.
Short-read Alignment
Although long sequencing reads produce more contiguous genome assemblies, their relatively high error rates make it difficult to generate a highly accurate final sequence. For this reason, it is advisable to apply correction and polishing procedures.
An effective strategy is to combine long reads with high-quality short-read data, such as that produced by Illumina platforms. This strategy is known as polishing with short reads, and to be carried out, short reads must first be aligned to the long-read assembly.
The goal of the read alignment is to map short sequencing reads efficiently to a large reference genome to identify the ‘correct’ genomic loci from which the read originated whilst taking into account errors in the sequence reads. The Burrows-Wheeler Alignment (BWA) algorithm is a well-known read alignment package that is based on a backward search with Burrows-Wheeler Transform (BWT), to efficiently align short sequencing reads against a genome sequence.
The BWA algorithm is part of the DNA-Seq Alignment functionality, that can be found under the Genome Analysis module. Data must be provided first, that is, high-quality short reads in FASTQ/FASTA format, and long-read assembly in FASTA format. Both, single and paired-end data are accepted. Additionally, OmicsBox allows adjusting many BWA options which are divided into three categories: algorithm options (e.g. minimum seed length, bandwidth…), scoring options (e.g. matching score, mismatching penalty…), and output options (e.g. minimum score, soft-clipping…).
Results are returned in SAM/BAM format. SAM stands for Sequence Alignment/Map format. In addition, a report and a chart provide information about the results, such as the number of mapped and unmapped reads of each input file.

DNA-Seq Polishing
As a final step, the short-read alignments are combined with the long-read assembly for polishing and error correction. Pilon is a fully automated, all-in-one tool for correcting draft assemblies, including very large insertions and deletions. Pilon works with many types of sequence data but is particularly strong when supplied with paired-end data from short-read libraries (e.g. Illumina). Pilon uses read alignment analysis to identify inconsistencies between the input genome and the evidence in the reads. It then attempts to make improvements to the input genome, including SNPs, indels, gaps, and misassemblies. For both, haploid and diploid genomes, Pilon produces more contiguous genomes with fewer errors, enabling the identification of more biologically relevant genes.
OmicsBox offers the Pilon algorithm through the DNA-Seq polishing application. Pilon requires as input a FASTA file of the genome along with one or more BAM files of reads aligned to the input FASTA file. The categories of issues to fix should be defined (e.g. SNPs, indels, gaps, local misassemblies…). Furthermore, there are some control and heuristic options that can be adjusted.
Pilon generates a FASTA file containing the polished genomic sequences. Pilon also returns a text file reporting all changes applied to the input sequences. In addition, a report and charts provide information about the results. For example, the proportion of each type of change that has been applied to the input sequences.
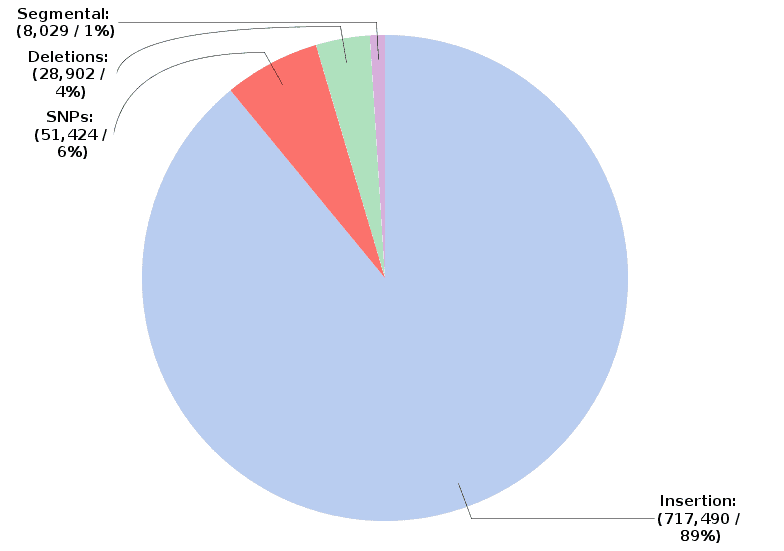
The strategy presented in this blog and fully implemented in OmicsBox allows to assembly genomes of any size with data coming from long and short-read technologies. This strategy ensures highly contiguous assemblies with a very low overall error rate.
References
- Kolmogorov M., Yuan J., Lin Y. and Pevzner PA. (2019). Assembly of long, error-prone reads using repeat graphs. Nature biotechnology, 37(5), 540-546.
- Li H. and Durbin R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics (Oxford, England), 25(14), 1754-60.
- Walker BJ et al. (2014). Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PloS one, 9(11), e112963.

