De novo transcriptome assemblies are required to analyze RNA-seq data from a species for which there is no reference genome. However, with the advancement of next-generation sequencing technologies, the amount of available sequencing data is growing exponentially. Because of this, assembly algorithms often generate a large number of transcripts. Removing redundancy from such data could be crucial for reducing storage space, computational time, and noise interference in some analysis methods.
A common procedure applied to reduce redundancy is the clustering of similar sequences. CD-HIT is a widely used program for clustering biological sequences to reduce sequence redundancy and improve the performance of other sequence analysis. Basically, CD-HIT is a greedy incremental algorithm that starts with the longest input sequence as the first cluster representative. Then it processes the remaining sequences from long to short to classify each sequence as a redundant or representative sequence, base on its similarities to the existing representatives. The similarities are estimated by common word counting using word indexing and counting tables to filter out unnecessary sequence alignments, which are used to compute exact similarities. It should be noted that the latest versions of CD-HIT implement a novel parallelization strategy and some other techniques to allow efficient clustering.
One of the algorithms in the CD-HIT package is the CD-HIT-EST algorithm, which clusters a nucleotide dataset into clusters that meet a user-defined similarity threshold, usually a sequence identity. This algorithm is suitable for clustering nucleotide sequences, such as those generated by RNA-Seq assemblers.
OmicsBox provides the Clustering functionality, based on CD-HIT. It can be found under the Transcriptomics module. To use it, the transcripts sequences must have been previously loaded on the platform. If the RNA-Seq reads have been not assembled yet, we recommend using the RNA-Seq de novo assembly application offered by OmicsBox (based on Trinity). If the transcripts have been previously assembled, they can be loaded into OmicsBox in Fasta format: File -> Load -> Load Sequences -> Load Fasta File.
Configure the clustering analysis is easy. Common CD-HIT-EST options are provided in two sections. In the “Algorithm Options” section, the way in which the sequence identity is calculated can be adjusted: the “global” mode computes the sequence identity as the number of identical bases divided by the length of the sorter sequence, while the “local” mode computes the sequence identity as the number of identical bases is divided by the length of the alignment. This sequence identity has to exceed the sequence identity threshold to consider that two sequences belong to the same cluster. Additionally, other options such as the bandwidth, the word length, the length cutoff… impact the way CD-HIT performs alignments. On the other page, the “Alignment Coverage Options” allow establishing sequence coverage thresholds, as well as unmatched sequence percentages cutoffs.
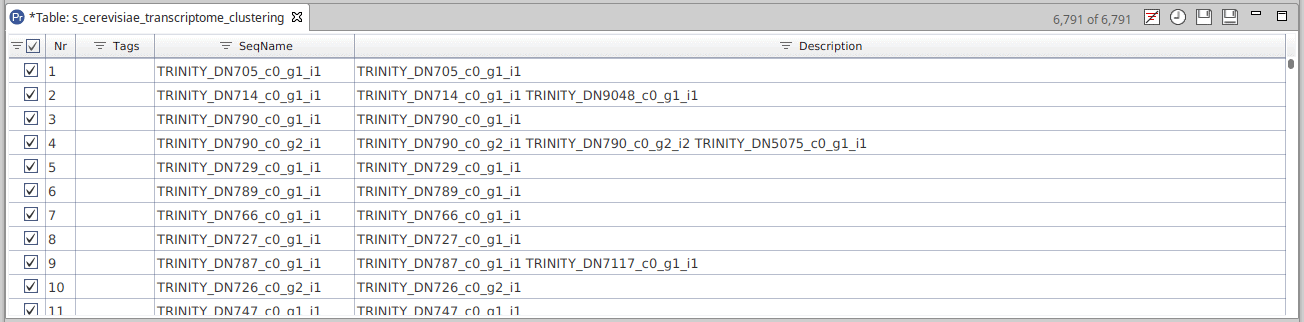
Once finished, results are returned in a project containing the representative sequence of each cluster. The SeqName field shows the identifier of the representative sequence. The Description field contains the sequence identifiers for the sequences that have been grouped into each cluster. In addition, the “Cluster File” is a text file generated by CD-HIT. It contains information about each cluster, such as the sequences grouped in each cluster, what is the representative sequence and how similar are the sequences between them.
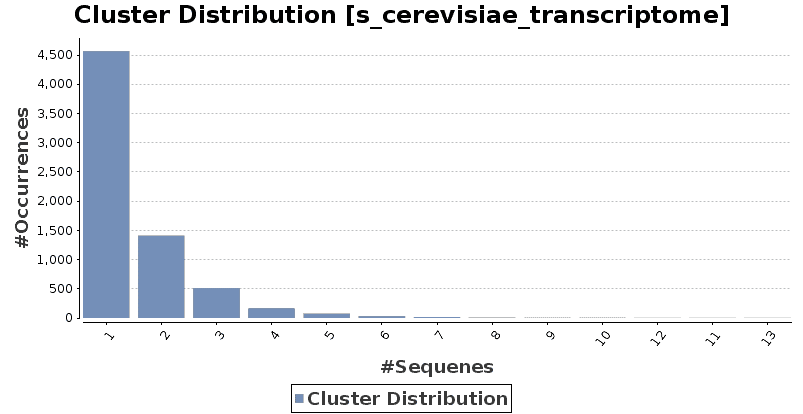
Finally, a report page and the cluster distribution chart will show the number of clusters of different sizes. Additionally, the list of the representative sequences of each cluster size can be exported.
Carrying out cluster on the RNA-Seq assembly results is a good practice. It allows reducing redundancy by clustering together very similar sequences. In this way, similar or misassembled sequences are discarded, and the sequences that are most likely to be real transcripts are recovered. Since it reduces the number of sequences, it is advisable to apply them before other procedures, such as functional annotation.
References
- Li W. and Godzik A. (2006). Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics (Oxford, England), 22(13), 1658-9.
- Fu L., Niu B., Zhu Z., Wu S. and Li W. (2012). CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics (Oxford, England), 28(23), 3150-2.

