Does RNA-seq-based Eukaryotic GeneFinding of Blast2GO require repeat-masking the whole genome shotgun (WGS) sequence? A case study in jute (Corchorus olitorius L., Malvaceae s. l.)
Debabrata Sarkar1, Carlos Menor2 and Nagendra Kumar Singh3
1Biotechnology Unit, Division of Crop Improvement, ICAR-Central Research Institute for Jute and Allied Fibres (CRIJAF), Nilganj, Barrackpore, Kolkata 700120, West Bengal, India. E-mail: debabrata.sarkar@icar.gov.in. ORCID iD: 0000-0003-3943-9646
2Blast2GO Team, BioBam Bioinformatics S.L., Avenida Peris y Valero 78-23, 46006 Valencia, Spain. E-mail: support@biobam.com
3Rice Genome Lab, ICAR-National Research Centre on Plant Biotechnology (NRCPB), Pusa Campus, New Delhi 110012, India. E-mail: nksingh@nrcpb.org
Introduction
Prior to gene prediction in eukaryotes, it is important to mask repetitive sequences including low-complexity regions and transposable elements (Yandell and Ence 2012; Ekblom and Wolf 2014). In the present study, we investigated the effectiveness of RNA-seq-based [WebAUGUSTUS (Hoff and Stanke 2013)] gene prediction using the Eukaryotic GeneFinding (EGF) module of the software Blast2GO (Conesa et al. 2005) with or without masking the whole genome shotgun (WGS) sequence of Corchorus olitorius L. (Sarkar et al. 2017). Since genome masking is not a prerequisite for EGF (in Blast2GO), our overreaching objective was to assess whether repeat-masking would improve the precision of protein-coding gene prediction and annotations using evidence from RNA-seq alignments as implemented in Blast2GO.
Material and Methods
The WGS sequence of C. olitorius cv. JRO-524 (GenBank accession LLWS01000000 vide BioProject PRJNA278717 and BioSample SAMN04160039) was used in this study. A repeat library was constructed, according to Campbell et al. (2014). In brief, MITEs (miniature inverted repeat transposable elements) and LTRs (long terminal repeat retrotransposons) were first searched with structural approaches using MITE-Hunter v11-2011 (Han and Wessler 2010) and LTRharvest (Ellinghaus et al. 2008)-LTRdigest (Steinbiss et al. 2009), respectively followed by the identification and collection of most repetitive sequences based on a de novo-approach using RepeatModeler v1.0.10 that uses RECON v1.08 and RepeatScout v1.0.5 (http://www.repeatmasker.org). The genome was soft-masked with all known and unknown repeats using RepeatMasker v4.0.7 (http://www.repeatmasker.org). The 454 RNA-seq reads of C. olitorius cv. Sudan Green (SRA accession SRR5145920 and GenBank TSA accession GFDJ00000000 vide BioProject PRJNA278717 and BioSample SAMN06199046) were aligned to masked as well as unmasked genomes using HISAT2 v2.1.0 (Kim et al. 2015) with default parameters. We used the Eukaryotic GeneFinding (EGF) module of Blast2GO v4.1.9 for RNA-seq-based (WebAUGUSTUS) gene prediction- with introns as hints from RNA-seq alignments- by selecting Theobroma cacao as the closest species of C. olitorius (Kundu et al. 2015; Sarkar et al. 2017). Complete genes were searched on both strands, without allowing in-frame stop codons. Basic gene prediction statistics were calculated by GenomeTools v1.5.9 (Gremme et al. 2013). Annotation features were retrieved as TxDb objects with the R v3.4.0 package GenomicFeatures v1.24.4 (Lawrence et al. 2013). Histograms were drawn using ggplot2 (Wickham 2009).
Results and Discussion
Using evidence from RNA-seq alignments, the EGF method of Blast2GO predicted 47,035 protein-coding genes from the unmasked genome (~377.4 Mbp) of C. olitorius cv. JRO-524 that covered 25,108 sequence regions, with a total length of ~209.4 Mbp (Table 1). When all known and unknown repetitive sequences including low-complexity regions and transposable elements (such as MITEs and LTRs) were masked, exactly the same numbers of protein-coding genes were predicted from identical sequence length and regions.
Table 1. Summary gene prediction statistics for masked and unmasked genomes of C. olitorius cv. JRO-524 (GenBank: LLWS01000000) annotated by RNA-seq-based (WebAUGUSTUS) Eukaryotic GeneFinding module of Blast2GO.
| Feature | Unmasked | Masked |
| Sequence regions | 25108 | 25108 |
| Total length (bp) | 209390614 | 209390614 |
| Genes | 47035 | 47035 |
| mRNAs | 47434 | 47434 |
| Exons | 217043 | 217043 |
| CDSs | 209879 | 209879 |
No differences were observed in the total numbers of mRNAs (47,434), exons (217,043) and CDSs (209,879) annotated in masked versus unmasked genomes. An examination of retrieved annotation features showed that distributions of CDSs, exons and transcripts per gene were identical between masked and unmasked genomes (Fig. 1). Similarly, distributions of CDSs and exons per transcript were uniform irrespective of repeat-masking prior to annotations. Our results thus establish that RNA-seq-based EGF module of Blast2GO is highly precise in predicting genes from non-model organisms without masking repetitive sequences in the genome.
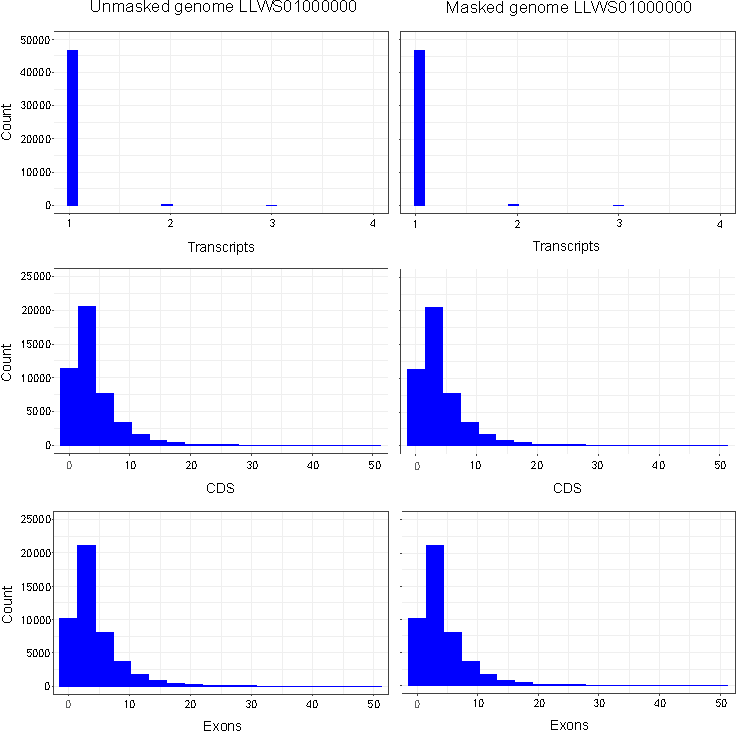
Figure 1. Histograms of exons, CDSs and transcripts per gene annotated in masked and unmasked genomes of C. olitorius cv. JRO-524 (GenBank: LLWS01000000) using RNA-seq-based (WebAUGUSTUS) Eukaryotic GeneFinding module of Blast2GO. Features were retrieved as TxDb objects with the R package GenomicFeatures
Conclusions
The Eukaryotic GeneFinding method of Blast2GO based on WebAUGUSTUS that uses RNA-seq alignments as intron hints can accurately predict and annotate protein-coding genes even when repetitive sequences remain unmasked in the genome. For masking all repetitive sequences in a genome involves memory-intensive lengthy computations that often require multi-core processors and/or a multi-CPU platform, Blast2GO offers an affordable approach for gene prediction without compromising the annotation results in a non-model organism with limited genomic resources.
However, we in no way propose to undermine the importance of masking repetitive sequences prior to gene prediction in eukaryotes.
Acknowledgements
We thank Director, ICAR-Central Research Institute for Jute and Allied Fibres (CRIJAF) for providing facilities. Financial assistance from ICAR-NPTC sub-project ICAR-NPTC-3070 is acknowledged.
References
Campbell MS, Law M, Holt C, Stein JC, Moghe GD, Hufnagel DE, Lei J, Achawanantakun R, Jiao D, Lawrence CJ, Ware D, Shiu S-H, Childs KL, Sun Y, Jiang N, Yandell M (2014) MAKER-P: a tool kit for the rapid creation, management, and quality control of plant genome annotations. Plant Physiol 164:513-524
Conesa A, Götz S, García-Gómez JM, Terol J, Talón M, Robles M (2005) Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21:3674-3676
Ekblom R, Wolf JBW (2014) A field guide to whole-genome sequencing, assembly and annotation. Evol Appl 7:1026-1042
Ellinghaus D, Kurtz S, Willhoeft U (2008) LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinformatics 9:18
Gremme G, Steinbiss S, Kurtz S (2013) Genome Tools: a comprehensive software library for efficient processing of structured genome annotations. IEEE/ACM Trans Comput Biol Bioinformatics 10:645-656
Han Y, Wessler SR (2010) MITE-Hunter: a program for discovering miniature inverted-repeat transposable elements from genomic sequences. Nucleic Acids Res 38:e199
Hoff KJ, Stanke M (2013) WebAUGUSTUS- a web service for training AUGUSTUS and predicting genes in eukaryotes. Nucleic Acids Res 41:W123-W128
Kim D, Langmead B, Salzberg SL (2015) HISAT: a fast spliced aligner with low memory requirements. Nat Meth 12:357-360
Kundu A, Chakraborty A, Mandal NA, Das D, Karmakar PG, Singh NK, Sarkar D (2015) A restriction-site-associated DNA (RAD) linkage map, comparative genomics and identification of QTL for histological fibre content coincident with those for retted bast fibre yield and its major components in jute (Corchorus olitorius L., Malvaceae s. l.). Mol Breed 35:19
Lawrence M, Huber W, Pagès H, Aboyoun P, Carlson M, Gentleman R, Morgan MT, Carey VJ (2013) Software for computing and annotating genomic ranges. PLOS Comput Biol 9:e1003118
Sarkar D, Mahato AK, Satya P, Kundu A, Singh S, Jayaswal PK, Singh A, Bahadur K, Pattnaik S, Singh N, Chakraborty A, Mandal NA, Das D, Basu T, Sevanthi AM, Saha D, Datta S, Kar CS, Mitra J, Datta K, Karmakar PG, Sharma TR, Mohapatra T, Singh NK (2017) The draft genome of Corchorus olitorius cv. JRO-524 (Navin). Genom Data 12:151-154
Steinbiss S, Willhoeft U, Gremme G, Kurtz S (2009) Fine-grained annotation and classification of de novo predicted LTR retrotransposons. Nucleic Acids Res 37:7002-7013
Wickham H (2009) ggplot2: elegant graphics for data analysis. Springer-Verlag, New York
Yandell M, Ence D (2012) A beginner’s guide to eukaryotic genome annotation. Nat Rev Genet 13:329-342
Note: Now it is possible to run Repeat-Masking in OmicsBox with the Genome Analysis Module. For more information please read the user manual.

