Barcoding and demultiplexing in high-throughput sequencing experiments
Barcoding, or indexing, is a widely used strategy in high-throughput sequencing experiments. This method enables the multiplexing of numerous samples in a single sequencing run by adding a unique DNA sequence to each sample before sequencing. During sequencing, the barcodes are read along with the DNA fragments, allowing the identification and split of sequencing reads belonging to each sample using demultiplexing tools like Cutadapt.
More Samples and Coverage At Lower Cost
The genomics field has advanced thanks to barcoding and demultiplexing, allowing researchers to increase the throughput of their experiments while reducing costs. Indeed, multiplexing has enabled the sequencing of large groups of samples in a single run. Which is particularly useful in large-scale studies such as population genomics, metagenomics, and single-cell genomics. The use of barcoding has also facilitated the sharing of sequencing data across different laboratories. It allows researchers to pool their data and analyze it collectively.
Analyzing multiplex sequencing data: First Steps
Quality Assessment
Analyzing multiplex sequencing data can be a tough challenge, but there are some crucial first steps to take. First, it’s essential to assess the quality of the raw sequencing data to identify any potential issues, such as base-calling errors or adapter contamination. This step can be performed using quality control tools like FastQC.
Reads Demultiplexing
Once the data quality has been checked, the reads need to be demultiplexed which involves separating the sequences into individual samples based on their barcode sequences. This step is critical to ensure that downstream analyses are performed on the correct samples or individuals. Various software tools are available to perform demultiplexing, and the choice of software depends on the sequencing platform, library preparation protocol, and sequencing data quality. Correspondingly, one of the most popular and versatile tools to demultiplex reads is Cutadapt.
Downstream Analyses
After demultiplexing, the reads can be aligned to a reference genome or assembled de novo to create contigs. These initial steps are important for generating high-quality data from high-throughput sequencing experiments. The correct selection of tools in these steps enhances the results from downstream analyses, such as variant calling, gene expression quantification, and metagenomic profiling.
Cutadapt is a fast and effective tool to demultiplex sequencing data
Cutadapt is a widely used tool for demultiplexing sequencing data that offers both speed and effectiveness. With its ability to quickly process large amounts of data, Cutadapt has become a popular choice for researchers looking to separate sequencing reads based on barcode information efficiently. The tool is also highly effective at removing adapter sequences, which can interfere with downstream analyses if left untreated. Again, cutadapt’s versatility allows it to use different sequencing technologies and experimental designs, making it a valuable asset for various research fields. Also, this tool is a reliable and powerful tool for demultiplexing sequencing data that can save researchers time and effort while ensuring accurate and reliable results.
Cutadapt in OmicsBox
- Demultiplexing with Cutadapt is included in the General Tools Module along with some other utilities to process sequencing data, such as FastQC, LongQC, and Trimmomatic.
- Initiating a Cutadapt run within OmicsBox is a streamlined process that facilitates rapid adjustment of demultiplexing to match the unique attributes of the data. The user just needs to provide the multiplexed reads (single or paired-end) and a text file containing the barcoding sequences (Figure 1A).
- As a result of the Cutadapt, OmicsBox splits the input sequences according to the barcodes that best match them. Moreover, it generates some matching statistics to evaluate the accuracy of the whole process.
- The OmicsBox implementation allows choosing how to split the matched sequences: By input file and barcode (Figure 1B) and only by barcode (Figure 1C).
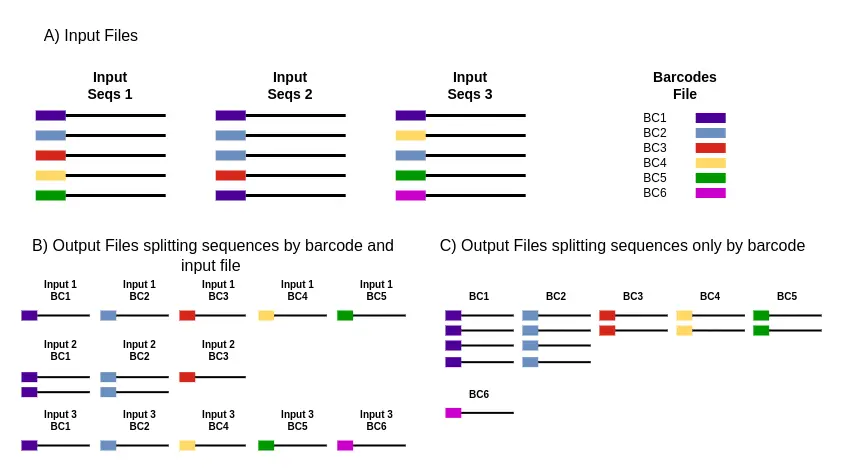
Figure 1. Inputs and outputs of Cutadapt in OmicsBox. Black lines represent the sequencing reads; colored boxes represent the barcodes. A) Input files: sequencing fastq files (1-3) and barcode files. B) Output files resulting from splitting the sequences by barcodes and input files. C) Output files resulting from splitting the sequences only by barcodes.
- OmicsBox allows efficiently combining this tool with some downstream analysis in the same workflow. For example, it is possible to quickly generate a workflow to analyze GBS data by combining Cutadapt with the Genetic Variation module (Figure 2).
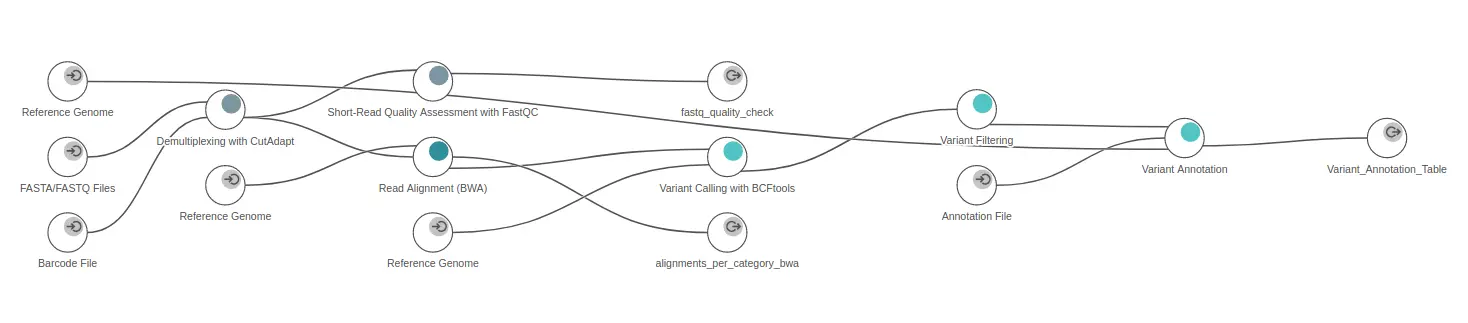
Figure 2. GBS workflow generated in OmicsBox.

References
Cutadapt removes adapter sequences from high-throughput sequencing reads.
Marcel Martin.
EMBnet. Journal, 17(1):10-12, May 2011.
Useful Links
About the Author

David Seide is a skilled bioinformatics professional with a BSc in Computer Science and a Master's degree in Bioinformatics. With over 10 years of experience, he currently holds the position of Head of Bioinformatics at BioBam.
In addition to his role, David serves as the Product Owner responsible for the Metagenomics module in OmicsBox, demonstrating his expertise and leadership in the field.

