RNA-seq technologies detect coding as well as multiple forms of noncoding RNA. RNA-seq can accurately measure gene and transcript abundance as well as identify known and novel features of a transcriptome. While the coding transcripts will lead to effector proteins, the non-coding transcripts are usually involved in the gene expression regulation and in the transcription and translation machinery.
In this evaluation, we will predict the coding potential for the transcripts of an RNA-seq experiment showing the different options of this tool which is based on the ‘Coding-Potential Assessment Tool 1 ‘ and an overview of the results.
Used dataset
To evaluate the overall performance and different analysis options of the Coding Potential Assessment Tool available in OmicsBox/Blast2GO we choose as test dataset the transcripts of Chr.1 of Equus caballus (horse) from the SRA database (NCBI). This dataset contains 455 transcripts assembled with Cufflinks from the transcriptome of equine peripheral blood obtained with Illumina HiSeq 2000 paired-ends.
Download:
Analysis steps
To perform the coding potential calculations, we select the Blast2GO project we will work with and chose the “Coding-Potential Assessment Tool” option under the “Tools” menu.
First of all, we need to select the accuracy of the coding potential cutoff. Two options are available:
- By default, the accuracy is calculated so that sensitivity and specificity are maximised in order to reduce the false positives and negatives.
- We can set a fixed accuracy, obtaining two cutoffs (a coding and a non-coding cutoff), allowing to classify more accurately the transcripts.
 Fig.1 – Coding-Potential Assessment Tool wizard.
Fig.1 – Coding-Potential Assessment Tool wizard.
 Fig.2 -Two graph ROC curves for automatic detection of
Fig.2 -Two graph ROC curves for automatic detection of
optimized CPS threshold and user specificity thresholds.2
To assign a to coding-potential value to each transcript the tool uses an empirical model built from coding sequences (mRNA and CDS) and non-coding RNA (ncRNA, lncRNA, snoRNA, etc). In the case, the number of available ncRNA sequences is less than necessary it is possible to use sequences from a higher, parent-taxa organism. Blast2GO offers a number of pre-built models for the most commonly used organisms as well as offers the option to create a species-specific model from the NCBI database. A third option allows to directly use local files if coding and non-coding sequences in fasta format to train a model.
To calculate the coding potentials of the “horse”-transcripts it is possible to use a mammal, pre-built model. However, we preferred to build a species-specific model with the sequences from the NCBI keeping the accuracy automatically calculated. Once the model has been created we observed an automatic accuracy of 0.939. Since the automatically assigned assurance seemed low for this model we decided to rerun the tool increasing the accuracy to 0.96. In this case, our sequences will be grouped in 3 categories: coding, unknown and non-coding.
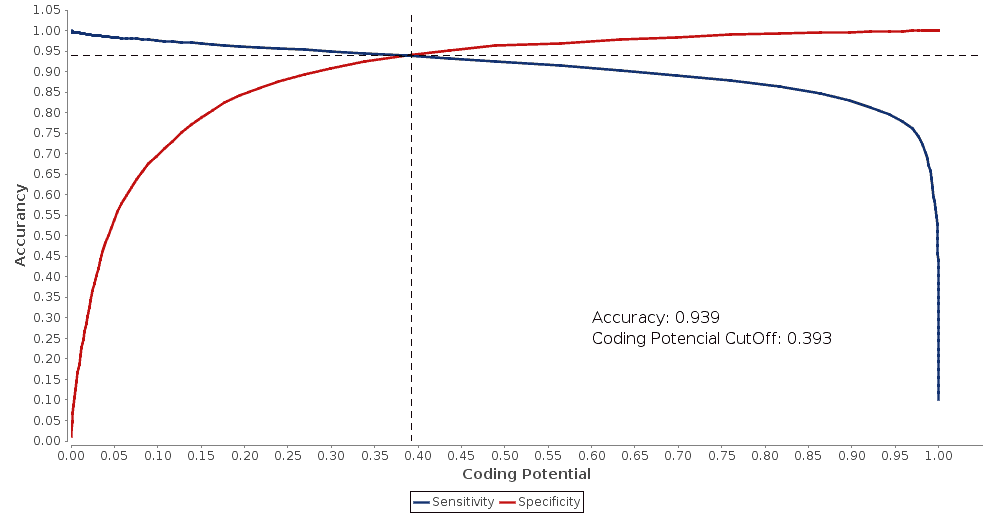 Fig.3 – The Double ROC curves with automatic accuracy. |
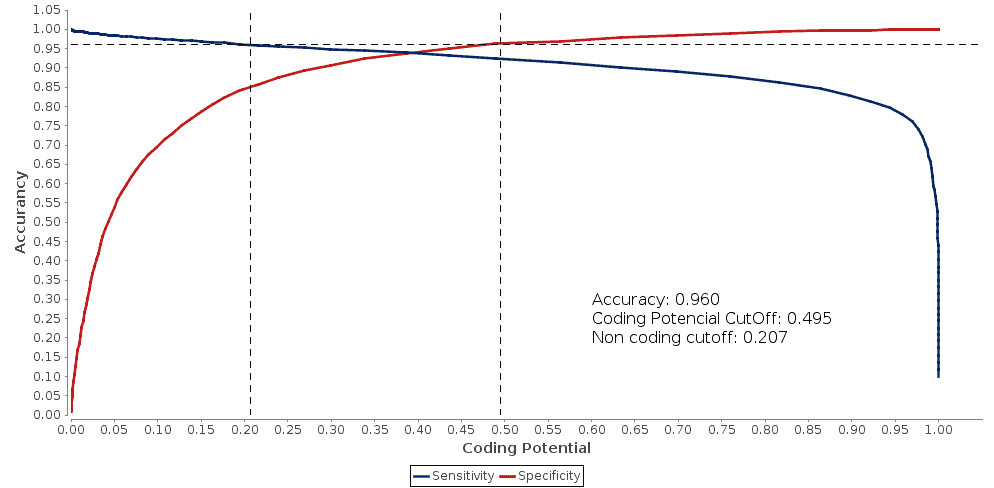 Fig.4 – The Double ROC curves with manually set accuracy. |
Assigning the accuracy manually we obtain two different cutoffs, a coding cutoff and a non-coding cutoff. With this two cutoff, we obtain the transcripts classified in three different categories:
-
 : if the coding potential is greater than the coding cutoff.
: if the coding potential is greater than the coding cutoff. : if the coding potential is less than the non-coding cutoff.
: if the coding potential is less than the non-coding cutoff. : if the coding potential is between the two cutoffs, the transcript is tagged as a transcript with unknown coding potential.
: if the coding potential is between the two cutoffs, the transcript is tagged as a transcript with unknown coding potential.
Performance
In order to evaluate the performance of this tool, we create a dataset of well-known protein coding sequences with the ‘Load sequences from BioMart’ tool, selecting as dataset the first 1000 coding genes of the human (GRCh38.p7) from the ‘Ensembl Genes 87. Once we have downloaded the sequences, we run the CPAT tool with the human prebuilt model, obtaining 99.1% as coding sequences. The sequences tagged as ‘non-coding’ have been blasted and revealed on his hits some a very unspecific Blast hit having a very small HSP with very high similarity.
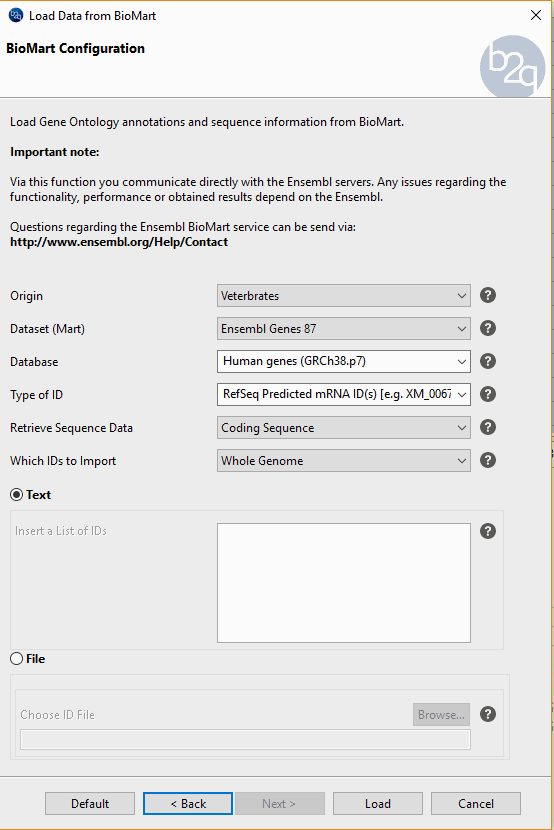 Fig.5 – BioMart tool used to import the sequences.
Fig.5 – BioMart tool used to import the sequences.
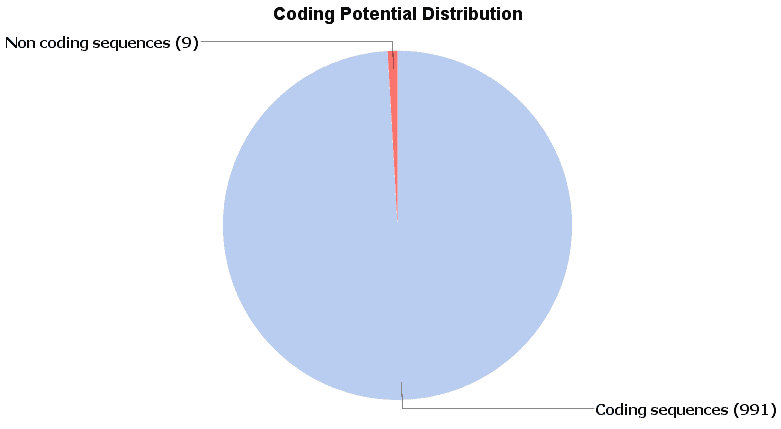 Fig.6 – Results of the coding potential on well-known protein coding sequences.
Fig.6 – Results of the coding potential on well-known protein coding sequences.
Results
Once the quality and precision of the model are adjusted, we can evaluate the prediction results.
On one hand, we obtain a “Coding Potential Table” which classifies each transcript with a tag and offers the values of the RNA length, the ORF length, the scores from where derive the coding potential score (Fickett score and Hexamer score) and the coding potential score itself. On the other hand, we can see a pie chart statistics of the coding potential distribution.
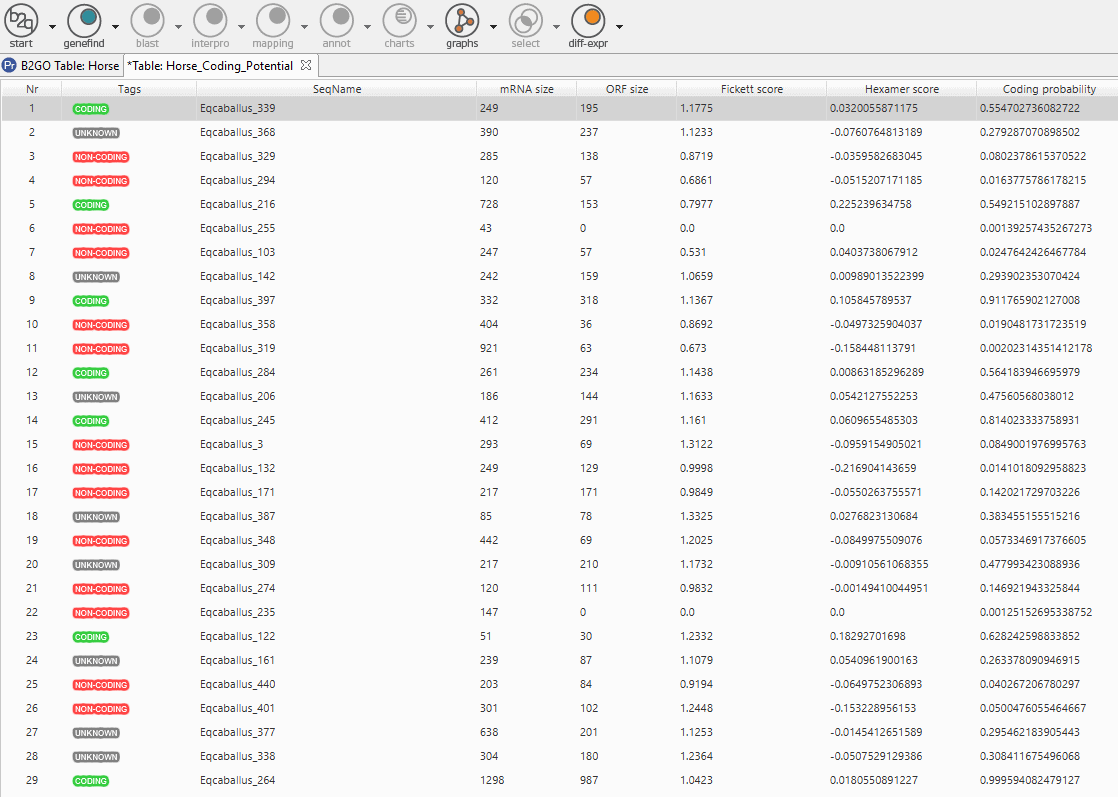
Fig.7 – Result Table
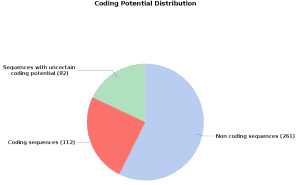
To further evaluate the classification transcripts have been blasted against a protein database. As expected, most of the non-coding results have also no Blast results while the coding ones do. However, we also observe transcripts classified as non-coding with Blast results – these might reveal gene expression regulators like ncRNA or miRNAs present in the protein database (also very unspecific Blast hits with a small HSP and high similarity could be aligned). These transcripts should be reviewed.
We also observe several transcripts classified as coding but without any Blast results. This could indicate that these potentially coding transcripts have not yet been added yet to any protein database and therefore might represent novel mRNAs.
Reference
1 Wang, L., Park, H. J., Dasari, S., Wang, S., Kocher, J. P., & Li, W. (2013). CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model. Nucleic acids research, 41(6), e74-e74.
2 Wucher, V., Legeai, F., Hedan, B., Rizk, G., Lagoutte, L., Leeb, T., … & Cirera, S. (2017). FEELnc: a tool for long non-coding RNA annotation and its application to the dog transcriptome. Nucleic Acids Research, gkw1306.

