Long-read sequencing technologies, led by Pacific Biosciences (PacBio) and Oxford Nanopore Technologies (ONT), have transformed transcriptomics research1. They enable scientists to study alternative splicing and isoform diversity in unprecedented detail.
Unlike short-read sequencing, which requires computational assembly of millions of fragments into transcript models, long reads can capture entire RNA molecules in a single read. However, due to various sources of noise, from biological variability to library preparation and sequencing, researchers must distinguish true transcripts from artifacts.
Numerous algorithms have been developed to tackle this challenge, yet no single gold-standard tool has emerged. Choosing the right tool depends on the dataset and specific research goals.
This post explains how to reconstruct and quantify transcriptomes using FLAIR2, one of the best-performing tools in recent benchmarks such as the LRGASP challenges3, and how to easily configure it within OmicsBox. As an alternative, OmicsBox also offers IsoQuant4, a similar tool with a distinct algorithm, which also enables similar workflows.
How FLAIR reconstructs transcriptomes
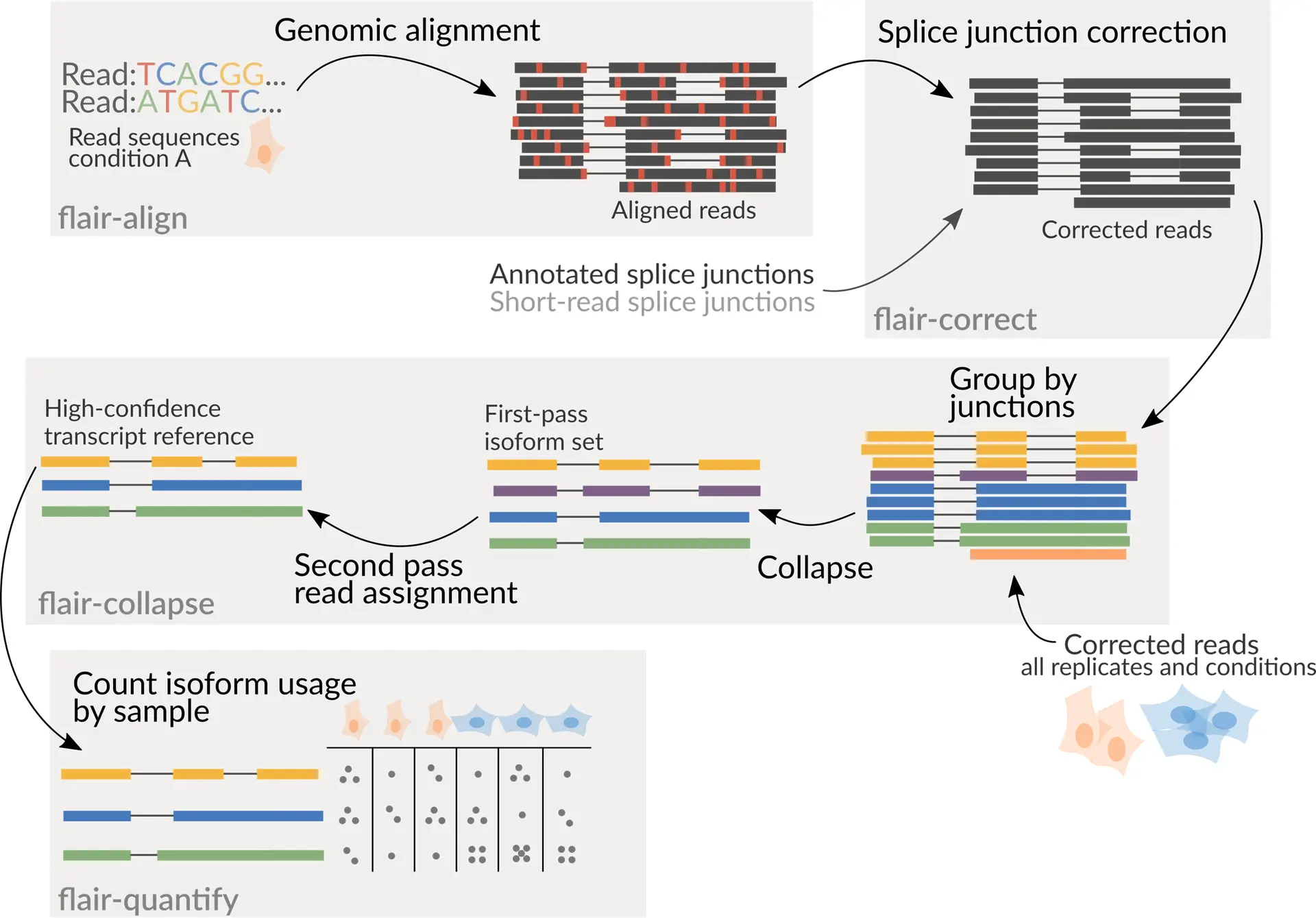
The FLAIR pipeline consists of four main steps (Figure 1):
- FLAIR-align: Align long reads to a reference genome with minimap2.
- FLAIR-correct: Correct splice junction errors using reference annotations and/or short-read data.
- FLAIR-collapse: Group reads by splice junctions and define common transcription start and end sites to generate unique transcript models.
- FLAIR-quantify: Map the original reads back to these transcript sequences to quantify expression levels.
Configuring FLAIR in OmicsBox
While manually setting up FLAIR can be complex, OmicsBox provides an intuitive interface that simplifies configuration and execution. Below is a typical workflow combining FLAIR for reconstruction and SQANTI35 for quality control.
Required Input Data
- Long-read RNA-seq data (PacBio or ONT) in FASTQ format, preprocessed according to platform guidelines.
- Reference genome in FASTA format.
- Reference transcript annotations in GFF3 or GTF format ,
and/or
short-read data in BAM or SJ.out.tab format
In order to perform transcriptome reconstruction from long-reads for non-model organisms where a reference genome is unavailable, refer to our blog post on the isON-pipeline.
Transcriptome Reconstruction
In OmicsBox, you can access FLAIR by navigating to
Transcriptomics → Long-Read Analysis → Transcript Identification → Identification and Quantification with FLAIR.
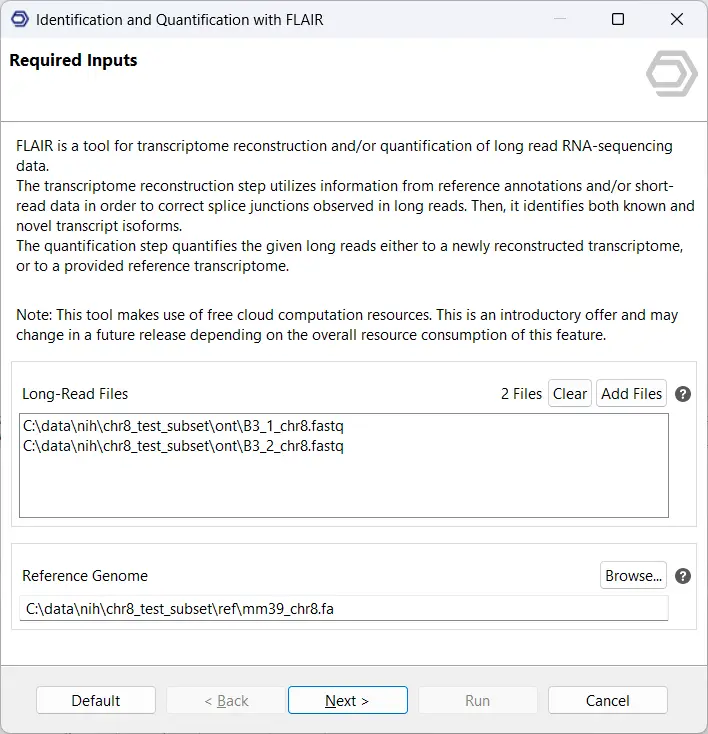
On the first page of the wizard, provide your long-read sequencing data in FASTQ format along with the reference genome in FASTA format (see Figure 2). These inputs are essential for FLAIR to align reads accurately and reconstruct transcript models.
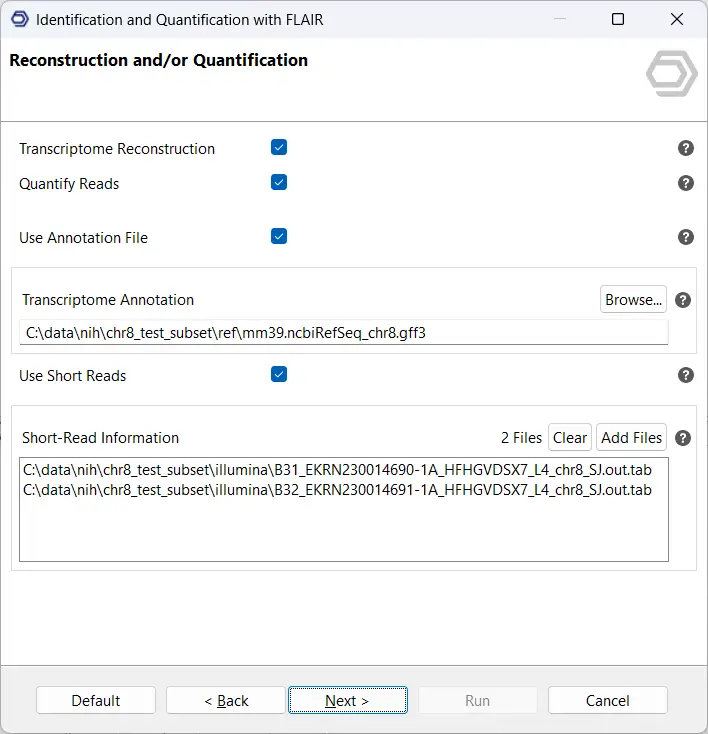
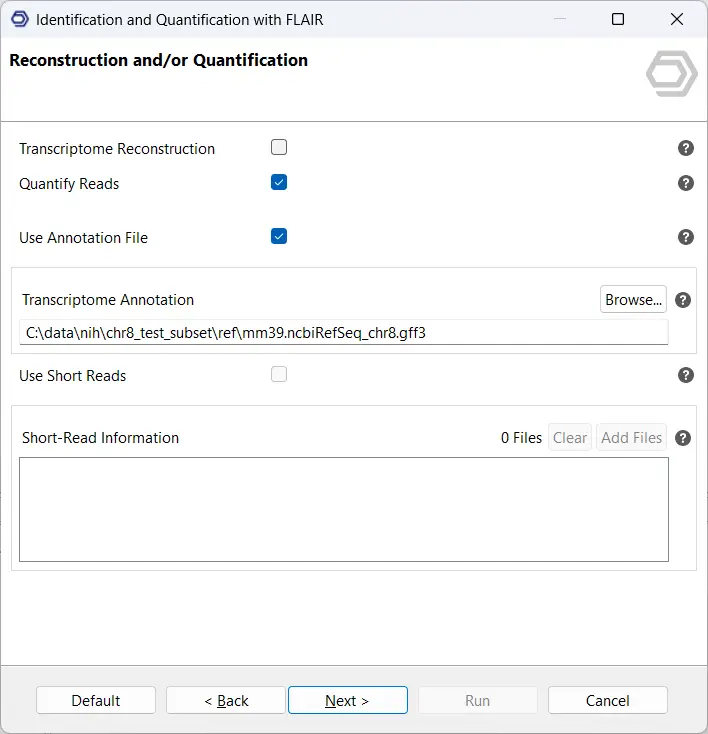
Next, select the option Transcriptome Reconstruction. On this page, you can provide a reference annotation in GFF3/GTF format, short-read RNA-seq data in BAM or SJ.out.tab format, or both (see Figure 3). Whenever possible, we recommend supplying both types of data. FLAIR uses reference annotations and short-read evidence to validate splice junctions and identify novel isoforms with greater confidence. Access to both data types improves the accuracy of the reconstructed transcriptome and enhances discovery of previously unannotated splice sites.
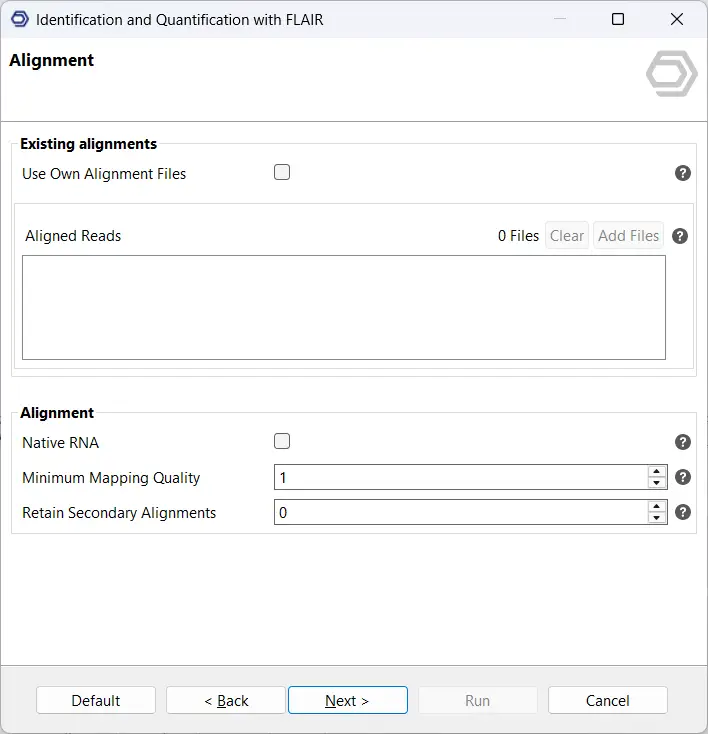
The next page allows you to choose how FLAIR performs read alignment. You can either provide pre-aligned reads in BAM format or allow FLAIR to handle alignment automatically using minimap2 (see Figure 4). Providing existing BAM files can save time and offers more control over the alignment process if you’ve already optimized your parameters. However, if you have not yet performed your own alignment, letting FLAIR perform alignment can be more convenient.
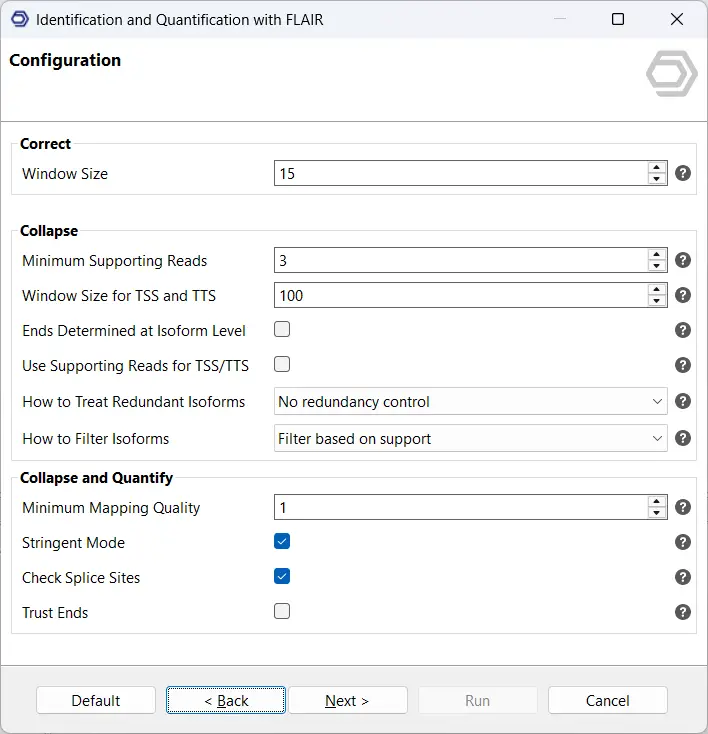
Finally, configure the parameters for the correct, collapse, and quantify steps (see Figure 5). These stages refine splice junctions, merge redundant isoforms, and compute expression levels. The OmicsBox user manual includes detailed explanations for each parameter. In general, the default settings perform well across most datasets. For well-annotated model organisms such as Homo sapiens or Mus musculus, we recommend enabling Stringent Mode and Check Splice Sites to increase annotation precision and minimize false-positive isoforms.
When the workflow finishes, FLAIR generates the following outputs:
- Transcriptome annotations in GTF format
- Transcript sequences in FASTA format
- A count table containing expression levels of detected transcripts
Transcriptome Curation with SQANTI3
After reconstruction, use SQANTI3 to curate and filter the transcriptome.
SQANTI3 compares reconstructed isoforms to reference annotations, flags potential artifacts, and retains only high-quality transcripts.
Provide SQANTI3 with:
- The same reference genome and reference annotation
- The GTF and count table generated by FLAIR
- Any available orthogonal data (CAGE peaks, polyA peaks, polyA motifs, and short reads)
If orthogonal data is available, enable SQANTI3’s machine-learning filter for optimal results. Otherwise, use the customizable rule-based filter.
SQANTI3 outputs:
- Summary plots visualizing transcript quality and novelty
- A curated transcriptome ready for downstream analyses
Re-Quantification on the Curated Transcriptome
Once SQANTI3 finishes curation, re-quantify reads using FLAIR on the filtered transcriptome.
This ensures the count table reflects only validated transcripts and reassigns reads previously linked to discarded artifacts.
To re-quantify:
- Open the FLAIR wizard again and disable Transcriptome Reconstruction on the “Reconstruction and/or Quantification” page.
- Provide the curated transcriptome from SQANTI3 as the input annotation (Figure 6).
FLAIR will skip the correct and collapse steps and directly perform quantification.
Alternatively, you can use this configuration to quantify reads against a standard reference annotation if you don’t want to discover new isoforms.
The output includes an updated count table suitable for downstream analyses such as differential gene or transcript expression analysis.
Conclusion
Long-read sequencing technologies provide an unprecedented view into transcriptome complexity. With OmicsBox, researchers can easily run the full FLAIR pipeline, from alignment and correction to curation and quantification, without manual setup or coding. This integration enables reproducible, high-quality transcriptome reconstruction and simplifies long-read RNA-seq analysis for both model and non-model organisms.
References
- Marx, V. Method of the year: long-read sequencing. Nat Methods 20, 6–11 (2023).
doi: 10.1038/s41592-022-01730-w - Tang, A. D. et al. Full-length transcript characterization of SF3B1 mutation in chronic lymphocytic leukemia reveals downregulation of retained introns. Nat Commun 11, 1438 (2020).
doi: https://doi.org/10.1038/s41467-020-15171-6 - Pardo-Palacios, F. J. et al. Systematic assessment of long-read RNA-seq methods for transcript identification and quantification. Nat Methods 1–15 (2024).
doi: https://doi.org/10.1038/s41592-024-02298-3 - Prjibelski, Andrey D., et al. “Accurate isoform discovery with IsoQuant using long reads.” Nature Biotechnology 41.7 (2023): 915-918.
doi: https://doi.org/10.1038/s41587-022-01565-y - Pardo-Palacios, Francisco J., et al. “SQANTI3: curation of long-read transcriptomes for accurate identification of known and novel isoforms.” Nature Methods 21.5 (2024): 793-797.
doi: https://doi.org/10.1038/s41592-024-02229-2

")
