The analysis of long-read RNA-sequencing data, such as from the platforms of Pacific Biosciences (PacBio) or Oxford Nanopore Technologies (ONT), can be rather complicated, and there are few well-established gold-standard tools in the field. This leads different researchers to choose different tools despite similar research objectives, which can lead to notable differences in their results.
One crucial step in such analyses is transcriptome reconstruction. The identification of both known and novel transcript models from long-read data is not just essential for transcriptome annotation itself, but can also have grave consequences on any downstream analyses, e.g., quantification and differential expression.
While benchmarks have attempted to assess different transcriptome reconstruction tools, none have specifically addressed how data from multiple biological replicates should be combined for this purpose.
In his poster for ISMB/ECCB 2025, BioBam team member and PhD student Fabian shares some early insights stemming from the preliminary results of his work trying to elucidate this common problem faced by many researchers.
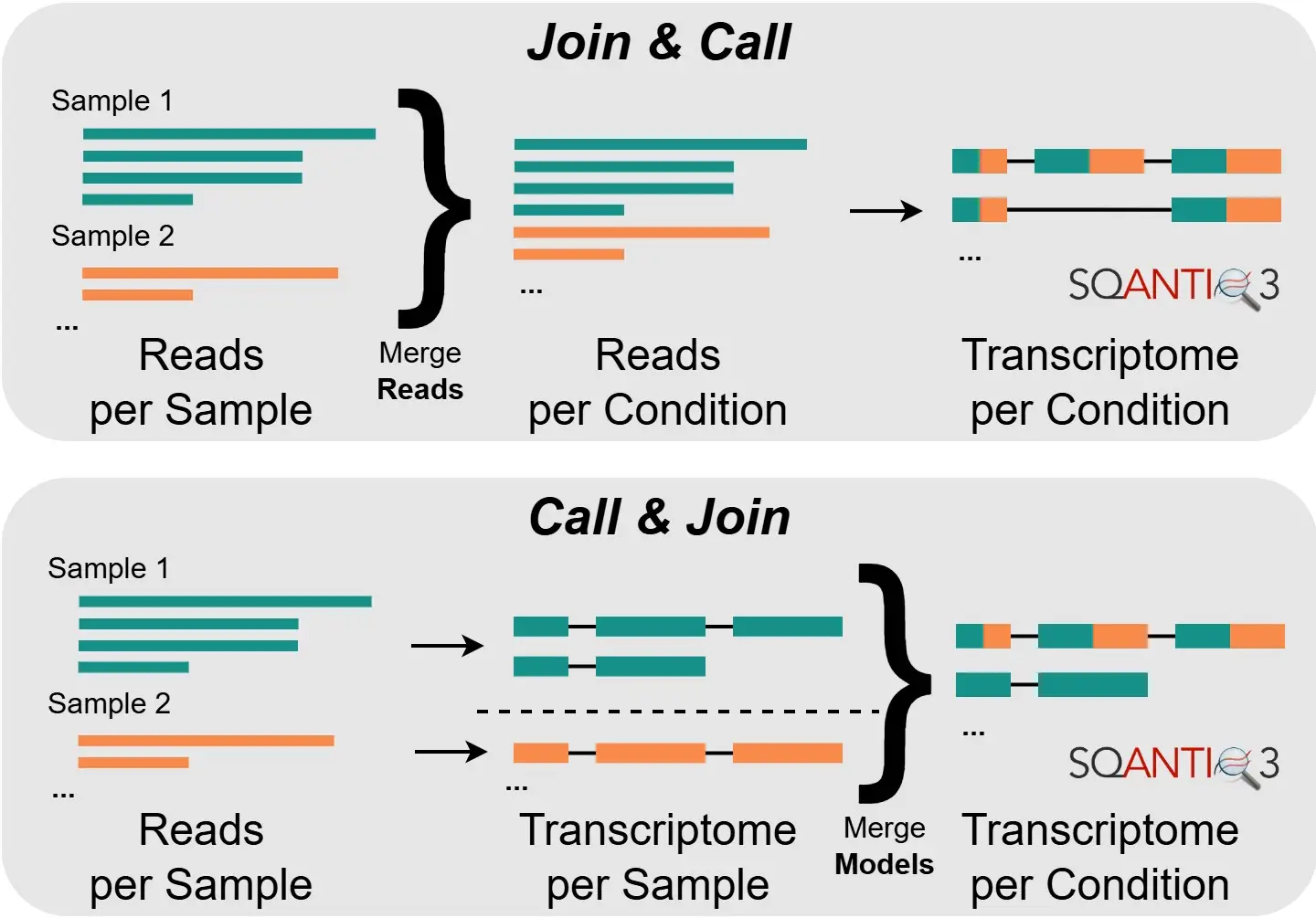
The first combination strategy examined, “Join & Call”, proposes to combine reads from all replicates before performing transcriptome reconstruction. This gives the process more power, especially in the discovery of rare, novel transcript variants.
The second strategy, “Call & Join”, follows the opposite approach: transcriptome reconstruction is first performed on each replicate independently, before combining the resulting annotations (using TAMA Merge). This not only requires fewer computational resources, but could also shed more light on sample-specific differences.
The preliminary results further show how different transcriptome reconstruction tools display different behavior between the two approaches. While some tools (e.g. FLAIR) appear to benefit from the “Join & Call” strategy, discovering notably more novel isoforms, other tools (e.g. IsoQuant) unexpectedly discover more novelty in the “Call & Join” strategy.
For this second class of tools, the same reads can be assigned to different unique junction chains across different samples This may mean that the increase in the number of isoforms does not actually represent an increased capability in discovering true novelty, but rather represents inconsistency in results across different samples. Such tools could therefore be unsuitable for use with the “Call & Join” strategy.
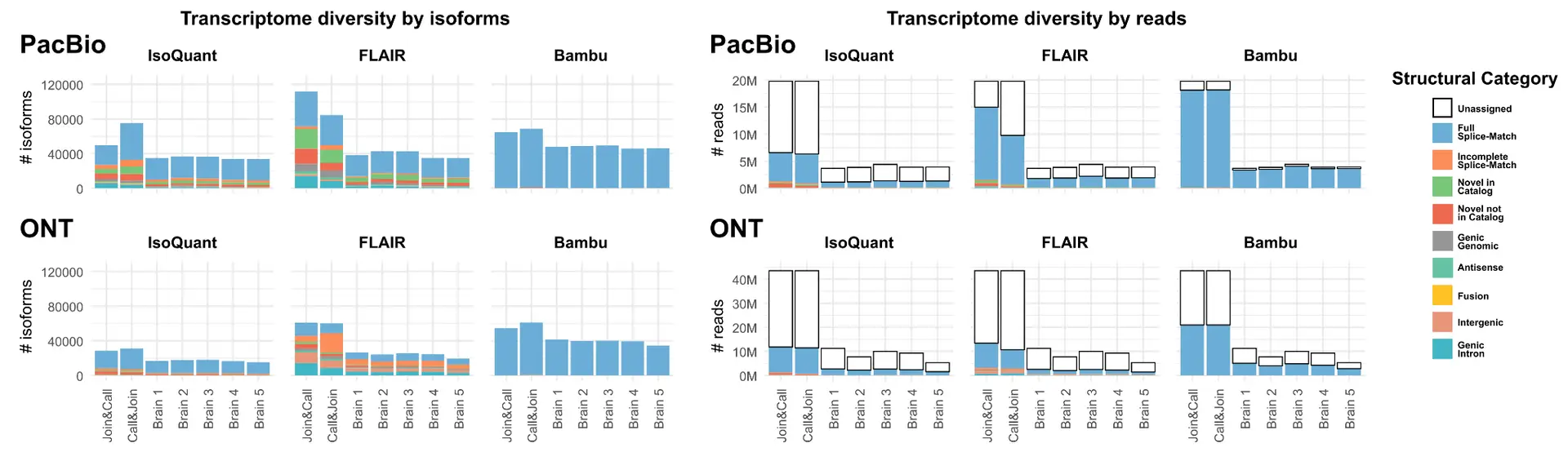
In conclusion, the poster recommends that researchers looking to analyze long-read RNA-seq data carefully consider their data type, research objectives, and computational resources when deciding on an approach for transcriptome reconstruction. Further, they should choose a combination strategy as well as tool jointly, as not all combinations of strategy and tool may be reliable.
For the study of rare, novel isoforms, the poster recommends using a tool that is more permissive in the discovery of novelty, such as FLAIR, with the Join & Call strategy.
Where the study of novel isoforms is secondary and/or computational resources are limited, the poster recommends using the Call & Join strategy with a more reference-faithful tool such as Bambu.
Read the full poster here.
About the Author

")
